e-mail :  ( Please write in ' Subject ' entry : ' METAPHYSICS ', in order for me to be able to distinguish your mail from spam )
( Please write in ' Subject ' entry : ' METAPHYSICS ', in order for me to be able to distinguish your mail from spam )
Having, in the previous document, presented an introduction and three Sections on the one-molecular view of the organism (Unimol), we now continue with Unimol, still largely following Oskar Müller, 1959. (See Note of author of website (from previous document) ) (and use the Page-back button of your browser to return to the present text)
Introduction (from the author of this website)
The present Section on the "renounced dimensions" is about the sizes of true molecules. If the organism is, according to the Unimol view, just one single molecule (embedded in an aqueous serum-like support medium), then this molecule, this organismic (i.e. living) molecule, must be of gigantic size (about the size of a whole organism) by far exceeding the size of the "normal" molecules (inorganic and organic) of chemistry, even exceeding by far the size of the so-called "biological macro-molecules". Is such a giant molecule really a true molecule? Is the conventional concept of "molecule" capable of including such giant "molecules", or must it be broadened? But, when so broadened, isn't then any macroscopic body whatsoever, - including true aggregates such as a granite, a solution, a suspension, - a molecule? Certainly not, because the constituents of a true molecule are not merely particles adjacent to one another, but constituents -- atoms -- chemically bonded to each other. And, according to aristotelian-thomistic metaphysics (see part XVe and part XVf of the present Series of documents), a molecule, any molecule, is not a mere aggregate, not a mere "many" of actually existing parts and particles, but a true whole, in which its constituents exist only virtually as subsistent particles (atoms), that is, they are not subsisting particles in the molecule but qualities of the molecule, i.e. regions or spots of a set of qualities of the molecule. In other words, the conserved and new qualities of the bonded atoms are now qualities of the molecule. And the same must apply to the giant living molecule. And as such the unimolecular nature of organisms guarantees their holistic nature. And, indeed, the present Section investigates (following Oskar Müller, 1959) whether or not true molecules can acquire macroscopic sizes (dimensions). It represents a defence in favor of the unimolecular status of organisms, a defence of Unimol. It must be noted that it is here not about the complete organism, which is, according to Unimol, a single molecule plus its multimolecular support medium, but about this single molecule only, which is the true "self" of that organism, supported by the mentioned medium. Being embedded in such a medium belongs to the overall strategy-to-exist of the organism, in fact of that molecule. But persistence of that molecule necessarily brings with it the necessary persistence of the support medium. The next support medium in which the living molecule plus its serum-like support medium is in turn embedded in the ecological niche of the relevant organismic species.
And again, the numbers in the müllerian text represent links to notes given (and so numbered) by Müller.
What thoughts one in fact has, until now, about the magnitude [the size] of those molecules constituting Life as a result of their cooperation in the "system" (as seen in the multi-molecular view of the organism)? Concerning this, we could not find precise indications. This is a very surprising and perhaps one-off fact in the domain of exact science.
One has departed from two fundamental presuppositions : 1. There exist larger physico-chemical units of molecular nature, being in some way protein-like or protein-simular, present as material substrates and foundations of Life. 2. Already the singular [biological] cell does consist of a large number of such molecules ( NOTE 328 ). Never, however, [the cell] consisting of one molecule only.
These two views are, of course, always right if one only considers the tremendous number of environmentally added and accessory small and large molecules [namely those of the support medium]. The true magnitude of the molecule was held to be varying between the order of magnitude of native biogeneous protein molecules (of colloid or crystallized nature) and an order of magnitude of a few (perhaps 3) powers of 10 lower [i.e. about a thousand times smaller] than the smallest known life-unit (also of the order of magnitude of virusses). As standard one still took the experimentally determined molecular weight of virusses and the stability considerations for macromolecules. The fact that one was satisfied with all this and never bothered, neither in thought nor experimentally, about specification -- which must be present, because it is about rationally natural things which are at our disposal in large numbers and which can be subjected to all kinds of methods including mental speculative ones, - that is a fact that can only be grounded by some As-If arguments.
A kind of rational foundation one had given, by saying that in such giant molecules, such as possibly genes and virusses, all methods of investigation point to the impossibility to set up determined molecular formulas or even only determine a molecular weight [roughly, how much heavier a molecule is with respect to a hydrogen atom] ( NOTE 328a ) (then at most indicating the order of magnitude or "degree of polymerization"). This is largely true, although the amino-acid sequence research of some still simple polypeptides for which one can also give a "precise" molecular weight, gives results which one can hardly interpret other than to represent a chemical structural formula and which at least expresses as much as the "structural" formulas of higher organic complexes which one as such sees as formulated, and this chemical structural formula anchors, in the general body of knowledge of the professional chemist, the still rather many, not expressed in the paper formula. [That is, in addition to the "paper formula" there is found, even in relatively simple compounds, a "formula" (a formulation) expressing "a many" not expressed by the paper formula. And this brings the simpler and smaller molecules closer to the giant molecules, or better, bring the latter closer to the former.]
But, anyway, does the methodical uncertainty of, for instance, the molecular weight and molecular unity, also mean abandoning and rejection of this state of affairs as to molecular weight and molecular unity? Certainly, there is no reason to do this. The precise formula is not so important anyway in this respect. One already abandons to determine one of crude rubber or of starch, without giving up the conviction that these macro-molecules further are wholly true uni-molecules. One also agrees to view a phenoplast as one single giant molecule ( NOTE 329 ). Even in virusses ( NOTE 330 ) and genes ( NOTE 331 ) one agreed to apply this concept, because it is useful and clarifying, and because to whose retention essentially nothing much stands in the way. Why one recoils at still higher units, for example chromosomes ( NOTE 332 ) and total-cytoplasm ?
If, in the domain of large-molecular substances [substances of which the molecules are of great size], the exclusive application of certain physical methods (barometric distribution of sedimentation [in centrifugues], osmotic pressure) is methodically indifferent as to distinguish between (1) true, i.e. molecularly disperse, solutions, and (2) colloidal mixtures or solutions, and therefore forbidding any methodical difference to exist between molecule and colloid system, and if, on the other hand, a chemical distinction along the line of the usual methods is difficult because such gigantic aggregates in many respects do behave like a system-mechanical association of the molecular-parts, - then one cannot, as a result, by far say that the impossibility to distinguish does prove that there is only the one kind of them, namely a colloid"system". If one would have seriously considered the reality of the other kind, then a series of conclusions could have been drawn from it, precisely covering many mysterious phenomena of Life (especially the problem of wholeness).
An unfathomable coincidence in the plan of scientific methodology -- perhaps under the sway of answering the enigmaticly inexaminable question-of-essence, an anwering applying not until at deeper levels -- had it that one passed over the evident solution of the problem. While, as to colloids, Wo. Ostwald expressed the concept : "world of the neglected dimensions", we may characterize the unimolecular life-system as the uniquely important world of not only the neglected or overlooked dimensions, but of the renounced dimensions.
The purely theoretically proceeding chemist seemed to have been forced, following earlier results, to deny, as to valence, the existence of many chemical compounds, which evidently actually do exist. The same applies to the mega-molecular forms ( NOTE 334 ). One thoughtlessly passes over the realized organismic mega-molecules and operates for good and bad with the classically sanctioned (so-called) macromolecules (the objects of macro-molecular chemistry), which one, inside the organismic, lets perform all those physical and chemical "tricks" that are necessary, in order to, limping and crutching, indicate precisely that, what could be realized directly through a deviating consideration [i.e. this deviating consideration, the Unimol view, will render all these chemical tricks understandable.].
That determinism of the precise molecular weight, of the stoichiometric synthesis and analysis, which is indispensable in the classical chemistry of small molecules because with it also the functional and qualitative is directly and inextricably connected, [that determinism of the precise molecular weight] does not play any role in the domain of the organismic [the domain of the living], and its experimental determination - if possible at all -- will be rather superfluous because it lacks to provide any special information. What is determinable in classical chemistry and necessarily leads to the functional and qualitative, can in organismic chemistry [the chemistry of organisms] only be concluded after the fact approximately from the exclusively observable functional-qualitative [aspect of the living organism]. That, of which it is, however, all about, namely the functional-qualitative, is in both cases there. The criterium of [chemical or otherwise] bonding can, as to be such and nothing else, be determined, and the molecular weight doesn't have any essential significance. Even in inorganic chemistry it is just about a, combined with other determinations, simple useful method of determination, which, however, already as a result of the often numerous isomers [= substances having the same formula of atomic proportions, and thus the same molecular weight, but with different structure of the molecule] of organic compounds, has no absolute significance anymore.
It is unclear why one objects so much against the evident total-bonding [continued, uninterrupted, bonding-sequence in an organism] which one should admit to, - as if it would contradict "something", as if it is impossible, as if it is something what one should not believe. Does this phobia for true organismic giga-molecules connect with a one-off "prohibition" of a synthesis (a continued conceptual connection) between inorganic micro-molecule and organism? It is as if with the recognition of organismic giga-molecules an unassailable and unconditionally to be maintained world replete with secured representations would collapse, whereas in so recognizing the organismic giga-molecule, we actually build up a new world of wonderful connections, a new world, supplementing and sensibly complementing the old one ( NOTE 335 ).
Where is the foundation excluding this assumption? It cannot be something empirical, for a chemist would certainly not be surprised that from the compound of individual molecules [a chemical compound having resulted from a chemical reaction between other such compounds] qualities and ways of reacting do originate, not directly derivable from the original molecules. [i.e. newly appearing features.]
It is not difficult to give the definition of a "supra-molecular complex" [i.e. a complex consisting of a number of individual molecules], but who can prove that biological objects, characterized as such complexes, will satisfy the chemo-physical aggregative definition sufficiently? [Müller here asks how one can actually prove that an organism is exclusively and wholly a system of (often interacting) individual molecules.]. How one wants to prove that non-usual molecules are no molecules at all? Did one have in mind, where the mysterious increasingly becomes more definite, a "uncertainty relation", in which simultaneously the transition between dead molecule and living system expresses itself?
Remarkably, nobody has proved that the living form is really a pure system of coordinated individual molecules. Cyto-chemically [the chemical nature of biological cells] one has abandoned Nägeli's micellar theory by reason of the at the time ignorance of effective macromolecules, but, strictly taken, one only has elevated this theory one level up, because one pseudo-micellarly aggregates organismic macromolecules in order to arrive at the functional morphological structures ( NOTE 336 ). Yes, even the 150 year old crude-optical impression of a structureless undifferentiated blastema ( NOTE of author of website ) was essentially continued, also after one had found an overwhelming multiplicity of submicroscopical structures, in which one most often has overlooked the fact that in this order of magnitude they can only be chemical structures ( NOTE 337 ).
One has set up the [conceptual] "construction" of [organismic] structures with the isolated [in the sense of having made free] extracted molecular ruin-material, having reduced the molecular order of size to the smallest extreme possible, and having assumed between the partners -- insofar as the all-capable "entelechies" [immaterial life-principles] and "life-forces" were excluded -- a relationship which to every professional researcher apparently lies in a domain alien to him. The chemist appears to have thought of physiological and histological moments, and the histologician of colloid-chemical ones. One spoke of mutual adherence [of particles] as if in the present domain there were another means of adherence than that what one indicates as -- possibly in a broadened but not overdone framework -- chemical bonding.
But if chemical bonding [is] in all, then homogeneous-molecular, and if homogeneous-molecularity, then whole-molecule. One might then say that the elements of this "proof" are of a suspiciously formal and verbal nature. But then one only needs to ponder about the practical utility and problem-solving ability of the conclusion, to find that it is overwhelming. The proper proofs may, it is true, not without further ado reveal themselves, but one now knows precisely where to look for them. One will find them, of this we are convinced, in great numbers, and then wonder that there was at all a time in which Unimol had a face of a mere fiction theory.
The, with the ascending hierarchy of forms, seemingly appearing new and higher regularities, apparently not derivable from the basic lower levels [now called "emergent properties or features"], can only be reduced to such lower levels by sharper definitional description and deepened detailed research. The remaining true qualitative rest then identifies itself with the [intrinsic] "hierarchy" of the molecule, [such] that namely with its growing in size, and thus with it to become atom-rich [consisting of many atoms], and therefore with its change, the qualities do change anyway. In organismic [living] molecules this is the case together with the peculiarity of an enhancement of specific qualities, a directed enhancement introduced as a result of differentiation and addition of potencies, an enhancement of specific qualities that partially seem to newly appear at certain [structural] levels or stages, but actually only stepping over a threshold, i.e. a conceptually (and sensibly) accessible to us threshold (such enhanced qualities being of the type : animation, consciousness, mind, etc. [i.e. true emergent features.] [So what is, already by many, ascribed to organisms, namely : the -- in addition to the conservation of certain properties of the organism's constituents -- appearance of new properties, not present in these constituents, not yet present at lower structural levels of the organismic body, indeed, the appearance of new, underivable properties of the organism, - is not a feature exclusively of organisms, but just a feature of molecules. It is an extended version of what we already knew about chemical molecules, and what has been extensively discussed in Part XVe and Part XVf. And indeed, in larger and larger molecules the appearance of new properties becomes more and more dominating. So in organismic, i.e. living molecules, we may expect, and indeed see, true emergent properties, properties only visible at the highest structural levels of the organismic body.]
It is not evident why an uninterrupted (continuous) chemical bonding in organismic living matter and a, based on that, possibly extremely high molecular weight (extreme, that is, with respect to that measured in inorganic small molecules), would transcend our power of imagination, a power that in the end certainly is not unseparably fixed to micro-molecules with their small number of atomic constituents. On the other hand, it is certainly not in the power of practical imagination to apprehend the organismic act as a sum-effect or cumulative property of a trillion of individual molecules.
Staudinger has pointed to the fact that biologists of earlier decennia rejected to work with macro-molecular substances [such as proteins], because almost all macro-molecular compounds are polydispersive [i.e. occurring in many variants [of dispersion]]. One wanted to relegate this domain to colloid-chemistry. Today we appear to have the same situation with the organismic, i.e. living, macro-[and uni-]molecular substances, which one apparently wants to relegate to theoretically proceeding botanists and zoologists. These then, surely, have a right to take them up, but methodologically these botanists and zoologists must at the same time be professional chemists.
Our view is not revolutionary as to the content with which it is concerned, but only with respect of the methodically conceptually formulated opinion about the content ( NOTE 338 ). Who doesn't know anything of molecules, to him the name may be indifferent ( NOTE 339 ). But he, or she, who does understand a lot about molecules is also able to overview the relevant criteria and can establish that our view is, at first sight, certainly astonishing (that is always so), but then becomes consistent and evident (that also is almost always so).
How, then, matters in fact are? One hardly has doubted that in the organism many of the familiar macro-molecules ( NOTE 340 ) have in turn come together in larger coherences (unspecific micelles, or specific functional units, and others), of which the element of bonding essentially was seen as to be mechanical (adhesive, crystalloid, macro-electrostatic, or other intermolecular forces), but where also true occasional [chemical] bonding was considered. Precisely in more recent [1959] literature one again and again does find such indications ( NOTE 341 ). Often one speaks with a certain degree of self-evidence about physiological mega-molecules having "bonds" with others, for which then in turn the same holds. But even the most diverse chemical knowledge apparently did not provide a stimulus to continue this possible line of enquiry all the way up to the very end. And probably one cannot start from here. We ourselves have arrived at our special view along a totally different line, which chiefly is a functional and genetic one, and which should not be seen as an immediate continuation and specification of the just mentioned view of possibilities, already because this [latter] view by itself may not be capable of explicitly lifting [relevant] thought out of the framework of the additive nothing-else-than-ensemble-system [view of the organism], and particularly that is absolutely important.
Of statements [about the molecular status of the organismic body or of its parts] that can be found in the literature we'd like to give, in addition to those mentioned in several Notes, a few examples.
H. Staudinger (1955) writes : ". . . It is to be noted that one single macro-molecule is still not living, how large and complicated it may be. For the state to be a living state, in addition to such a macro-molecule still many smaller molecules are needed, molecules that are with precisely prescribed chemical reactions working together in a specificly formed environment, and in this way [together with the macromolecule] representing an "atomos" of the living [body] [a body, being, according to Staudinger, composed of several such macro-molecules and their companions.], which atomos, precisely as is the case of the atoms of the elements, is indivisible, otherwise it will lose its living-state. . . . Atoms of organisms = cells, for instance bacterial spores . . . consisting of several hundreds of differently-sized macro-molecules (molecular weight [M.W.] = 104 - 106 ) and several 100 000 small molecules . . . [now describing a still higher structural level]. Remarkable [is] the relative small number of species of organisms . . . and the definite and strict heredity of the generations, despite the most diverse possibilities of construction [of a body] and ensemble [of constituents] . . . So the constancy [and therefore definiteness] of the [organismic] species must essentially be conditioned [and restricted] by still further form-factors also guiding the further development . . . into a determined direction . . ."
Here, undoubtedly, we have to do with a terse and rightly felt description of the system, in which only -- as everywhere -- two things are lacking : the essence [or nature] and the chemical overall-bonding, with which its goes over into the organismic system. At the same time (1955) Staudinger has said that an upper limit in molecules of macromolecular substances cannot be given. So to our assertion that macromolecules are not the building blocks of the living cell, but these themselves being macromolecules, nothing revolts in this respect [and we may extend this line towards Unimol].
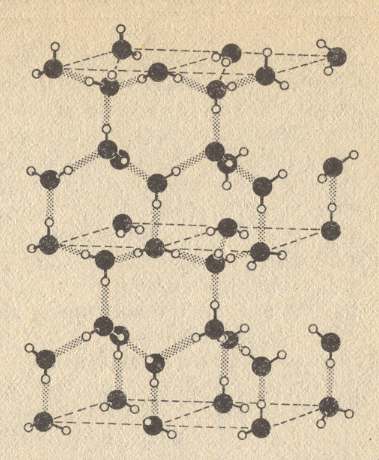
Frey-Wyssling (quoted by K. Mühlethaler, 1957 ) feels unequivocally enforced, on the basis of his microscopical studies, to depict the omnipresent fibrillar structures in a bonding fashion [i.e. as bonded to one another]. Globular macro-molecules, aggregating linearly, must have two pre-formed places (poles, "points of adhesion"), on their surface, onto which [places] adjacent particles adhere to one another. In planar and three-dimensional arrangements correspondingly more points or planes of adhesion are present. ( Frey-Wyssling had expressed his assertion that the basic plasm [the protoplasm] consists of large thread-like protein molecules knitted by their points of adherence into a three-dimensional framework, already at a time in which the semi-liquid basic cytoplasm [plasma of the cell interior outside the nucleus] was still viewed as optically empty. The meshes of the framework are filled with a continuous -- continuous like the molecular frame [itself] -- liquid, i.e. aqueous, phase, containing salts, carbohydrates, lipoids, etc.). As those things realizing the bonds, Fr.-W. discusses various bonds-by-cohesion, covalent bonds, and hydrogen-bridges.
Conjectures about the maximal length of bonding-sequences we have not found. But if such sequences are present, then they must extend "transversely" through the whole cytoplasm. Fr-W's protoplasm is in principle built up uni-molecularly. In one particular attempt we cannot, however, follow him : namely in establishing molecular types of building blocks, whose nature may imply the most diverse morphological forms. This certainly is true in many circumstances, leading, however -- as a way of thinking [because it is reductionistic] -- away from the most important [Every molecule, also the largest, we must view as a holistic entity]. We are at the same time suspicious that, in all correctness of the basic idea, precisely this material framework cannot be stable thermodynamically and chemo-kinetically, and will [have to be] alter[ed] already purely externally (Moreover, it is of course a mechanically [because of the alledged building-block construction] constructive morphological-only element lacking the essence).
The acute neurophysiologist W. Bargmann, it is true, says : " The specific organic foundations of the cell structures are the protein bodies with their large molecules . . . with other cell components having come together into ... large molecular complexes . . . in which by whatever careful addition of all chemical compounds that can be extracted from cells one cannot constitute a living mass." W.B., however, also characteristically speaks of "mega-molecular structures, revealed to us by the electron microscope, as well as of the research into the molecular constitution of the cell" ( The living cell nucleus shows a vague, the dead cell nucleus a distinct framework).
W.I. Schmidt's (zoologist, polarization-optician) ideas about the molecular constitution of the cell boil down to this [bold face mine] :
Space-lattice-like arranged thread-like [fibrillar] molecules do constitute with their micellar structure a kind of submicroscopic skeleton of the protoplasm. In the meshes of this framework the other cell-ingredients are deposited.
[again, bold face mine] The proportion of high-molecular to low-molecular compounds in an organism or in a tissue has been estimated by R.Signer (1955) as to about 20 : 1. The autor also mentions the physico-chemically conditioned ability of large molecules to bond to one another by supra-molecular forces, and the ability to localize also smaller molecules. Therefore, the large molecules become substrates for a spatial-temporal order of biological structures out of which the morphological builds up, an order that is hardly apprehensible even today.
These examples may suffice. They show how much one, except for naming ( NOTE 342 ), everywhere grants the qualitative rationality of the unimolecularity [of organisms, or at least of cells]. What always catches our eye are the "molecules" [said to be] bonded to one another chemically and regularly (and thus practically also stoichiometrically, at least when referring to parts). [So one not only speaks about atoms bonded to other atoms (resulting in molecules), but also often of molecules bonded to other molecules.]. It [i.e. saying "molecules bonded to other molecules"] is unequivocally meaningful and practically convenient (and used by us in the very usual way), if one means with it moments qua reaction, moments qua-generation, analytic moments, or merely a simplified naming (of the type di-phenyl, [di-peptide], and perhaps also glycyl-glycine, and others). With it, however, one should not neglect the status of substances-so-named as new qualitative one-forms [That is, by naming a given substance after some repeating molecular element (such as a repeating group of atoms in the molecule) in it, one should not forget that such a substance is not merely the sum of these repeating molecular elements (plus some other, non-repeating elements). It is a new substance (chemically and metaphysically).].
We shall now interrupt the present Section "The world of the renounced dimensions", being about how large certain molecules can actually become and so enquiring into the possiblity of unimolecularity in organisms. We interrupt it in order to insert something of the theory of proteins, especially their chemical structure, i.e. the precise nature of their molecules, and the ability of these molecules to chemically connect with one another, forming (in organisms) still larger complexes. And because the living molecule must be more or less protein-like, and because proteins are chiefly made up of amino-acids bonded to each other by the "peptide bond", it is very important to study this bond in particular.
For all this, we shall quote a number of paragraphs from John Gribbin's book "In search of the double helix", 1987, which is about the discovery of the DNA machinery in organisms and all what went before. In that book some elements of the theory of proteins are presented in a clear way and are important in discussing Unimol. And, of course, after the conclusion of this insertion, we will return to "the world of renounced dimensions", continuing with Oskar Müller's discussion of Unimol.We will not in all cases literally quote parts of the text of Gribbins's book. We will just explain parts of the theory of proteins by making use of this text.
Resonance
The backbone of protein molecules is formed by the concatenation of amino-acid residues resulting in a so-called polypeptide chain. In forming such a chain the amimo-acid residues are connected with each other by the so-called peptide bond. This bond originates as a result of a "condensation reaction" between the COOH group (the carboxyl group) of one amino-acid and the NH2 group (the amine group) of another amino-acid. Normally, a molecule containing the COOH group acts as an acid by readily giving up the hydrogen atom in a chemical reaction, releasing it as a hydrogen ion (H+), but, when involved in the formation of the peptide bond, the COOH group, under suitable conditions, gives up, not a H, but an OH group, and here thus the COOH group acts as a base in a chemical reaction. In fact, the structure of the carboxyl group is best thought of as a resonance between two alternative forms [the acid and the base form]. It is, therefore, important to precede the theory of the peptide bond with the theory of resonance, and we can do this most conveniently by considering the example of the ozone molecule (O3).
The principle of resonance says that if a molecule can be described in two (or more) equally acceptable ways (where "acceptable" effectively means states with the same energy, different versions of the lowest possible energy state for that molecule), then the molecule has to be thought of as existing in both (or all) of those states simultaneously. The 'real' molecule is a hybrid of all the possible structures with the same lowest energy. The example of ozone, a relatively simple molecule, gives an idea of what is going on, and is especially interesting because it brings out other features of chemical bonding at work.
The oxygen atom (O) has its outer (second) shell occupied with six electrons, and as such the whole atom is neutral [ 8 positive charges (protons) in the nucleus, 2 negative charges (electrons) in its first shell, and 6 negative charges (electrons) in its second and last shell]. The most stable state for this atom is an electronic configuration such that its outer shell contains eight electrons (then the atom has the inert gas configuration of its electrons). So when it manages to actually gain two electrons, by sharing two pairs with other atoms (one pair-member of its own and one pair-member of the other atom), its outer shell is 'full' (now having 8 electrons), meaning that an oxygen atom can form two covalent bonds with other atoms (See for chemical bonding : previous document).
So oxygen has a valence of two, with six electrons in its outermost occupied shell. It is easy to write down (ignoring the inner, completely filled electron shells) representations of molecules such as water, H2O, and oxygen, O2, on the picture of shared electrons :
O==O
Each bond, indicated by a dash, here : "--" (in most figures we simply write : "-" ), represents a pair of shared electrons. But how do you want to account for the fact that oxygen also forms a tri-atomic molecular form, ozone, O3? The first step is to appreciate that covalent bonding is not the whole story, and that the ionic transfer of an electron from one atom to another (as it happens also in pure ionic bonding) ought also to come under consideration, as it is also in the hydrogen molecule ( H2 , H +H - ). One way this could happen is for one oxygen atom to give up an electron to another oxygen atom. The first is left with a net positive charge and an outer shell containing 5 electrons. It now has the capacity to make 3 covalent bonds (instead of 2). The other oxygen atom has gained an electron and a net negative charge. It has now room for only one more electron (to complete its outer shell) in a covalent bond [i.e. it can only make one covalent bond with another atom], it has an outer shell of 7 electrons. If we follow the convention of using a dash to represent a pair of electrons in a bond, and dots to represent the electrons [in the outer shell] that are not part of the covalent bond, this gives us the two following ways to represent the structure of an ozone molecule :
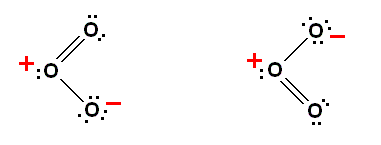
Diagram above : the ozon molecule (O3).
Left image : The oxygen atom on the left has given up one electron taken up by the lower right oxygen atom. Therefore, the left oxygen atom now has only 5 electrons in its outer shell, and may, consequently form three covalent bonds with other atoms (by sharing electrons). Here it is so bonded with a double bond to a second oxygen atom and with a single bond to another. The lower right oxygen atom has gained one electron and thus now has 7 electrons in its outer shell. So in order to complete this shell it can form only one covalent bond. And indeed it is so bonded to the first atom, i.e. with a single bond. In order not to confuse things : The lower right oxygen atom (still in the left image of the diagram) has seven electrons in its outer shell (indicated by half the dash representing the bond [one electron of its own] and the six dots, each representing one electron not taking part in bonding. And because an electrically neutral oxygen atom has six electrons in its outer shell, the present atom, having seven electrons in its outer shell, has a net negative charge of one unit, indicated in the diagram with a minus sign.
Right image : The oxygen atom on the left here has given up an electron, not, as above, to the lower right oxygen atom, but to the upper right oxygen atom.
Spectroscopic techniques, which measure the amount of energy radiated or absorbed by a molecule, provide a direct measurement of how much energy is stored in each bond. A double bond is stronger than a single bond. It provides a tighter link between atoms and holds them closer together. So, if this picture is correct, there ought to be evidence of two different bond energies [in one and the same individual molecule] corresponding to two different bond lengths [rendering the molecule asymmetric], apparent in the spectrum of ozone. There isn't. Instead, the spectroscopic studies show quite clearly that ozone molecules are held together [i.e. the three atoms of each molecule are held together] by two equal bonds, each equivalent to a bond strength of 1.5 [rendering the molecule symmetric]. The explanation is that the 'real' structure is a resonance between the two possibilities outlined in the diagrams, a hybrid structure like the hybridization which gives us the sp3 orbitals in the carbon atom. (See previous document, the chemical bond). Unlike that kind of hybridization, however, in this case the picture does involve a re-arrangement of electronic charge which produces asymmetry (in one and the same molecule). One atom has effectively lost an electron, and has a resulting surplus of positive charge in its vicinity. The other two atoms have each effectively gained half an electron, with a corresponding increase in the negative charge at either side of the molecule. Weak net charges of this kind commonly arise in molecules, especially large molecules which contain many atoms. Because opposite charges attract one another, while like charges repel, this leads to a tendency for large molecules to stick together in certain ways, and, indeed, for different bits of very large molecules to stick together in a weak form of electrostatic bonding. This turns out to be of great importance in the molecules of Life. The possibilities are almost endless.
Amino-acids, the peptide bond, proteins, and macro-molecules.
Even at the atomic level, the living world is different from the world as a whole. There are 92 chemical elements which occur naturally on Earth. Only 27 of these are essential components of living things, and not all of these are essential to all living things [essential to all living things are C (carbon), O (oxygen), H (hydrogen), and N (nitrogen), the "organogeneous elements" ]. Furthermore, the proportions of these atoms of Life found in living things are not the same as the proportions in which they exist over the Earth as a whole. Leaving aside water, which makes up more than three-quarters of the weight of most living things, more than half the weight of your body (its "dry weight") is carbon, a quarter is oxygen and nearly 10 percent nitrogen. For comparison, about 47 percent of the Earth's crust is oxygen, 28 percent silicon (Si), and some 8 percent aluminium (Al), combined together to make rocks. Biologically important atoms have been selected by evolution because of their chemical properties -- essentially, the way they form chemical bonds -- which in turn give biomolecules the special properties of Life. Most biomolecules are compounds of carbon. The other element strikingly over-represented among living things is nitrogen.
In terms of dry weight, proteins are by far the most important molecules of Life, dominating the make-up of your body. And proteins contain about 16 percent of nitrogen, an even higher proportion than in the body as a whole. Many proteins are very large, complex molecules, but like all biomolecules they are built up from simple units and subunits.
At the next step up from individual atoms, the basic building blocks of Life are units, consisting of a few atoms, which [units] are frequently found in more complex structures. These [units] include molecules such as ammonia (NH3) from which one hydrogen atom may be lost to form an amine group (-NH2) joined to a carbon chain :
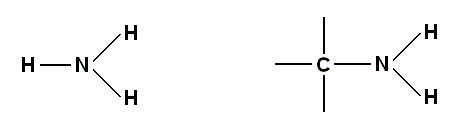
and the carbon atoms themselves, as well as forming benzene rings [rings of six carbon atoms], often crop up in many other configurations, a few of which are shown below, where the unlabelled bonds may join up with any other group R, such as the amine group, another carbon chain, or a carboxylic acid group, the latter denoted by -COOH. The structure of the COOH group can be represented either like this :
emphasizing the importance of the single hydrogen atom at the end of the chain, or like this :
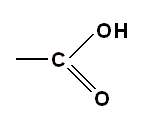
indicating that the OH group acts as a unit in some chemical reactions [for instance in forming the peptide bond between amino acid (residues) ].
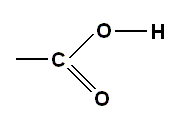
The simplest definition of an acid is a substance that readily gives up a hydrogen ion which combines with an OH (hydroxyl) group from another substance (base) to give water, H2O. The carboxyl group (COOH) acts in this way in many chemical reactions, which is why it is also called the carboxylic acid group, and it is this group which provides the acidity in amino acids, and in other organic acids. One of the simpler members of the carboxylic acid family is acetic acid, which is the principal ingredient in vinegar :
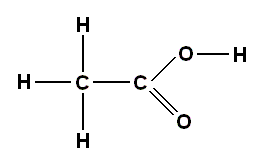
Confusingly, however, the carboxyl group can also act as a base, under suitable conditions, giving up the OH group in a chemical reaction. And it is this reaction, not its acid nature, that is involved in the formation of the peptide bonds which hold amino acids together in chains.
In fact, the structure of the carboxyl group is best thought of as a resonance between two alternative forms :
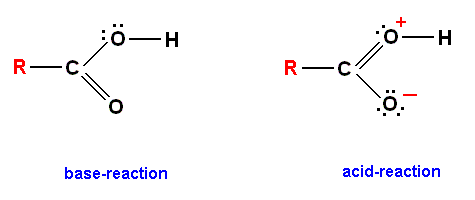
Diagram above : The two resonating versions of the carboxyl group (COOH).
In the right hand version of the carboxyl group an electron from the upper oxygen atom has been transferred to the lower oxygen atom. Therefore, in the vicinity of the upper oxygen atom (having given up one electron) we have a net positive charge of one unit, while in that of the lower oxygen atom (having gained an electron) we have a net negative charge of one unit. (Of itself the oxygen atom has six electrons in its outer shell, so when it gives up one it becomes positively charged, and when it gains one it becomes negatively charged.)
In this right-hand version of the carboxyl group the upper oxygen atom and the carbon atom share two pairs of electrons, constituting the double covalent bond between that oxygen atom and the carbon atom. Two of these electrons (in each pair one) originally belong to that upper oxygen atom (in its outermost shell). Further, the outermost shell of this same upper oxygen atom contains two electrons not involved in bonding. Finally, in this same atom, an electron pair is shared with hydrogen, one member of that pair originally belonging to the oxygen atom (in its outermost shell). So the outermost shell of the upper oxygen atom of the right-hand version of the carboxyl group contains 2 + 2 + 1 = 5 electrons, which is consistent with the fact that it has given up one electron from its outer shell.
The lower oxygen atom of that same right-hand version of the carboxyl group originally possessed, in its outermost shell, one of the electrons in the electron pair shared with carbon. It further has, also in its outermost shell, 6 electons not participating in bonding. So in all, this oxygen atom has 1 + 6 = 7 electrons in its outer shell, consistent with the fact that it has gained one electron, rendering the atom to be negatively charged.
The upper oxygen atom of the left-hand version of the carboxyl group originally has, in its outer shell, one electron from the pair shared with carbon, and one electron from the pair shared with hydrogen. In addition it has, also in its outermost shell, 4 electrons not participating in bonding. So that makes up for this oxygen atom a total of 1 + 1 + 4 = 6 electrons in its outermost shell, and therefore this atom is electrically neutral.
The version on the left [of the above diagram] contributes 80 percent of the character to the bonding structure, the version on the right 20 percent. The quantum properties of the electrons which make up the bonds are crucially important in determining the overall structure of more complex molecules which contain this kind of group, and in determining details of the way the group reacts chemically. But when writing out approximate structural formulas for more complex molecules which include the carboxyl group it is usually easier to use the shorthand COOH, with the detailed structure of that particular group taken as read. In the same way, chemists commonly use the shorthand forms of other common groups, such as -NH2, -CH3, and so on.
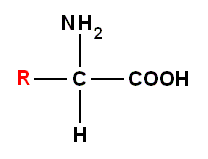
Very many amino acids can exist in principle -- and many have been made in the lab -- but only 20 are found in [natural] proteins. The 20 amino acids presented in the next Figure are the building blocks of Life. They are present in all proteins.

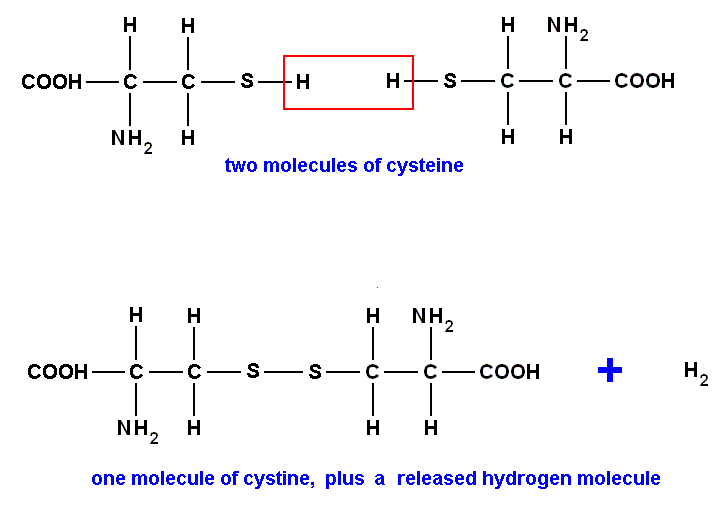
In addition, there are two amino acids that are found in a very few proteins, and one of the common amino acids, cysteine, very readily combines with another molecule of cysteine to make cystine :
More concisely, since the group
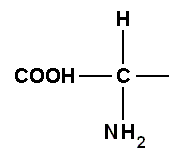
is the same in all amino acids, we can ignore it and concentrate on the side chains which distinguish one amino acid from another :
combine to make :
The link between the two half-molecules is a disulfide bridge [an S-S bridge], and is very important to an understanding of the structure of proteins. It is, however, to some extent a matter of choice whether you regard cystine as a different building block of Life, or simply another version of cysteine. So you may find different sources referring to 20, 21, or 23 vital amino acids, or simply, more cagily, to "about 20". We shall stick with the 20 shown in the above Figure. [The disulfide bridge can also connect two cysteine aminoacid residues belonging to different proteins, and so hooking up these latter two, resulting in one larger molecule. The disulfide bridge may also connect the two arms of a folded protein chain and so stabilizing its, for example, hairpin structure.].
Even before the last few essential amino acids were identified (threonine in 1938) it had become clear that they link up to make proteins by forming polypeptide chains. This is just about the simplest way that amino acids can get together, and the groundwork for this understanding was laid by the work of the German chemist Emil Fischer in the first decade of the 20th century, although in fact the proof that proteins are polypeptide chains, and not more complex structures, came only in the 1930s. The chains -- like polymers -- are formed by condensation, releasing a simple [in the sense of one single] molecule as two amino acids join together. The amine group attached to one amino acid gives up a hydrogen atom, while the carboxyl group on the other amino acid gives up a OH group. A new bond -- the peptide bond -- forms between the -CO on the end of one amino acid and the -NH on the end of the other, to make a dipeptide, a union of two amino acids :
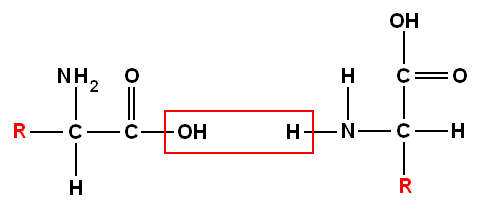
combine to produce :
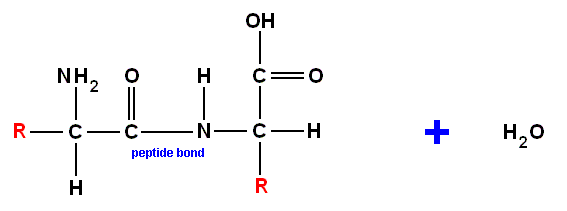
Notice that the two groups R need not be the same, and that the new molecule is bent. In fact, of course, all the carbon bonds are angled [when single, the four bonds extend from the center of an (imaginary) tetrahedron (in which the carbon lies) to its vertices] in the way Pauling explained in terms of hybrid orbitals [Pauling was a key theorist of chemical bonding]. We just draw some of the chains in straight lines for convenience. The protein backbone is formed by the concatenation of many amino acids by peptide bonds. In spite of the extra COOH and NH2 groups in side chains, proteins do not contain branches with peptide bonds extending from a pendant NH2 or COOH group. A living organism requires fibers but not cross-linked resins. Cross-linking occurs, though, with hydrogen bonds, but these are much weaker than peptide bonds.
This dipeptide itself now has a COOH group on one end and an NH2 group on the other. Each of them can combine with the appropriate group on the end of another amino acid to lengthen the chain further, and in the same way those new ends to the chain make further links, making a polypeptide. Just like in polymers, the result is a zig-zag chain. But in this case the spine of the long molecule is formed by alternating carbon and nitrogen atoms (two carbons, one nitrogen, two carbons, one nitrogen . . .). Starting at some arbitrary point in the chain, we have one carbon atom which has both a single hydrogen atom and the characteristic group [denoted as R] of a particular amino acid attached to it as a side chain. This carbon atom is bonded to another carbon atom which has an oxygen atom attached to it [by a double bond], and that carbon atom is bonded to a nitrogen atom [by the peptide bond], which in turn attaches to the next carbon atom that carries a complex side group [denoted as R]. As has been said, the groups denoted by R need not be the same. So the [general] pattern repeats. The general structure is like this :
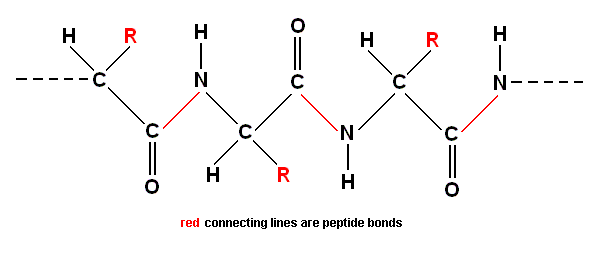
One of the key features of this polypeptide chain is that the peptide bonds form a rigid structure, held firmly in place by quantum mechanical resonance. It was this that gave Pauling a clue to the way protein chains coil up. But we can bring out the most important feature of the protein chain by ignoring the zig-zags and looking just at the main spine with its side groups attached. The carbon atoms in the main chain which carry the amino acid side chains are labelled with the Greek letter alpha [We here label them with the word "alpha" instead of with the letter itself.], to distinguish them from the carbon atoms which form part of the peptide bonds between adjacent amino acid components :
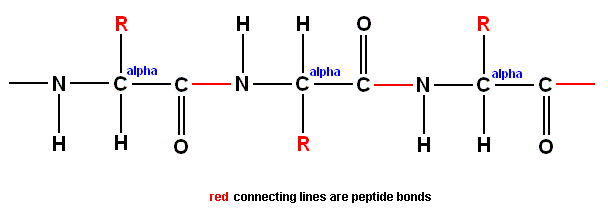
Even more simply, in a purely schematic representation taking no account of the geometry at all, we can indicate the structure of the molecule by putting all of the amino acid side chains on the same side of the spine :
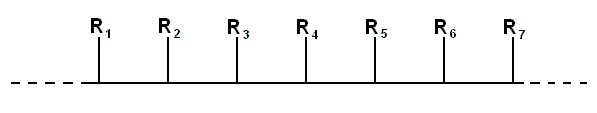
Displayed like this, it is easy to see how the chain carries a message -- the "words" are the side chains, the residual bits of the amino acids that make them different from one another. It is no mystery that a long string of such "words" conveys a biologically important message.
Among other things, it was found that the C-N bond in the peptide link [thus not the other C-N bonds in a polypeptide chain] is shorter than it 'ought' to be, and that, thanks to a quantum resonance, it has a partially double-bonded character, like the resonant bonds in the benzene ring. This makes it impossible for the protein chain to rotate around these bonds, and holds the whole peptide linkage [i.e. the atomic group in volved in the peptide bond] flat, like this :
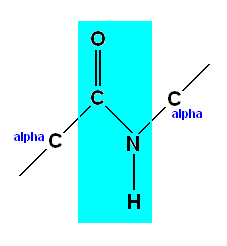
Both of the two bonds attached to an alpha atom, however, are free to rotate. It is as if the chain has two flexible linkages, then one rigid one, then two more flexible joints, and so on in a repeating pattern.
The polypeptide chain twists at the mentioned locations in order to place the successive O=C-N-H planes at the proper angles for maximum stability [= minimum energy]. These angles are limited [for reasons of stability] to a few combinations in order to avoid steric interaction of R groups on the amino acid units with C=O groups and with each other.
A major factor in determining the conformation of the polypeptide chain is hydrogen bonding [see previous document in the Section "the chemical bond"]. The O=C-N-H group [in which the C-N forms the peptide bond] acts as both acceptor and donor [of an electron], and certain arrangements of the chain are stabilized by extensive hydrogen bonding between the C=O group of one amino acid and the N-H of another unit. Two important types of conformation are a helix, which is stabilized by hydrogen bonds within a chain, and a pleated sheet arrangement with hydrogen bonding between adjacent polypeptide chains.
The most common helical structure is a right-handed spiral [as it is curved around an imaginary cylinder] in which every C=O group is perfectly positioned for hydrogen bonding with the N-H group three units away and above it in the spiral. This conformation is called the alpha-helix. A complete turn around the axis contains 3.6 amino acid units. The C=O and N-H bonds, and the hydrogen bonds between them, lie along the surface of the "barber pole", almost parallel to the axis. The R groups are tipped outward, with minimum interactions. The alpha-helix provides a readily extensible, elastic chain. Wool fiber and also the fibrous contractile protein of muscle are largely alpha-helix structures.
It is the ability of the alpha-helix to accommodate varying R groups, that stabilizes this secondary structure relative to many other conformations. Glycine units, each one being an amino acid without an R group [but with a single H instead] [see last entry in the left-most column of the above Figure], can, of course, also be accomodated [in the structure], but their presence [in a polypeptide chain] lessens the advantage of the alpha-helix compared to other possible arrangements [i.e. glycine units do form the helix (H-bonds between their C=O and NH groups), but the absence of R groups lessens the (mechanical, or otherwise) advantage of its being an alpha-helix.] . The cyclic amino acid proline [above glycine, in the above Figure], with a rigid ring between the N and alpha-carbon [when going, in the proline molecule, from its N-atom to its alpha-atom (i.e. the C-atom connecting to the COOH group), we move in a "circle"], cannot be accommodated in the alpha-helix and disrupts the helical structure wherever it occurs in the chain. [The above paragraph was taken from MOORE and BARTON, Organic Chemistry : An Overview, 1978.]
Since 1951, a great deal more research has confirmed the nature of the helix often found in fibrous protein. In fact, it is rare for the helix to dominate the entire length of a protein chain. Folds and cross linkages alter the overal shape of many proteins, and there may be stretches dominated by the alpha-helix, as well as straight stretches and other portions held by different cross linkages even along the length of a single polypeptide chain. [In order to get more understanding of Unimol] it is surely worth the trouble to look at some of the ways in which simple linkages between alpha-helices themselves can explain some of the structure of our bodies, and of other animals, and why keratin can make substances which are superficially as different as hair and tortoise shell.
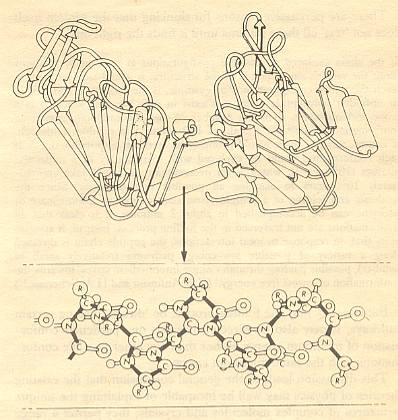
The geometry of the alpha-helix arises because the family of proteins that together make up the keratins, contain amino acids that neatly fit such a structure, and do not, by and large, contain amino acids that would distort the helix. In addition, those helices contain a lot of cysteine residues, the groups that are capable of forming disulfide bridges between polypeptides (see Figure above). In hard keratins, such as tortoise shell, or horn, the helical coils lie side by side, joined to each other by many disulfide bridges as indicated in the next Figure :
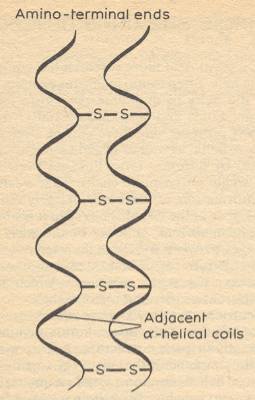
Figure above : Alpha helices, while themselves stabilized by hydrogen bonds, can be joined to their neighbors by disulfide bridges. (After GRIBBIN, 1987)
[From the point of view of Unimol, and also from that of aristotelian-thomistic substance-metaphysics, we must say that the alpha helices so bonded with each other result in a new molecule, result in a new Substance in the metaphysical as well as in the chemical sense.]
These bridges are true covalent links, an thus much stronger than hydrogen bonds. So the result is a sheet of keratin molecules held firmy together [of course it as such is one single molecule or even a part of it]. Add more sheets above and below, and it is easy to see how Nature builds up the structure of a tortoise shell, or your own fingernails.
But what about your hair?
In fibers of hair, the strength of the bonding between individual alpha-helices is again the disulfide link. But in this case sets of three alpha-helices are twisted together, like the strands of rope, to make up a supercoiled triple helix. The disulfide bonds hold the three members of each triple helix tightly together, and the three helices all run in the same direction, in the sense that their amine groups are all at the same end of the rope. Eleven of these three-stranded ropes are bundled together to make one hair microfibrile, and hundreds of microfibriles are bundled together to make one hair [whether the three-stranded ropes are in turn connected to each other by chemical bonds (S-S, or H-bonds) to form a microfibrile, I do not know, but it seems evident. And, further, whether these microfibriles are connected to each other to form a hair by chemical bonds also seems evident.]. Once again, even without going into details, it is straightforward to see how the properties of human hair are related to the sub-microscopic properties of the protein molecules.
It is also straightforward, on this picture, to understand something of the hairdresser's art. If hair is treated with a chemical compound that breaks up the disulfide bridges, the connections between the individual strands in the triple-helix ropes will be weakened. The hair will become soft and easily manipulated, and can, for example, easily be curled into a new shape. Then, when you have made the curls, all you have to do is wash the hair with another chemical, one that attracts hydrogen out of the cysteine residues (forming cystine, see Figure above) and allows new sulfide bridges to form between the adjacent alpha-helices in their new configuration. Take the curlers away and the hair stays 'set' in its curly shape, thanks to the new disulfide linkages. Pauling and Corey's masterpiece of chemical deduction can, among other things, explain the 'permanent wave'. The phenomenon depends on the nature of the chemical bonds between sulphur and hydrogen atoms, and Pauling explained chemical bonding in terms of quantum physics. The 'permanent wave' is a phenomenon of quantum physics.
What of the beta-keratin structure, distinguished by its unique X-ray fingerprint? That, it turns out, is not a helical structure at all. Instead of the polypeptide chains coiling around in helices, they form zig-zags very much more like the Figure above. And instead of the hydrogen bonds forming between different atoms on the same chain, they form, in much the same way, between the equivalent atoms in adjacent chains. The result is a structure which is superficially somewhat like the structure of a hard keratin, but in which the links between chains are formed by hydrogen bonds, not disulfide bridges. So the whole thing is much softer and more flexible -- indeed, one [kind of] fiber which has this pleated sheet structure, as it is called, is silk. See next Figure.
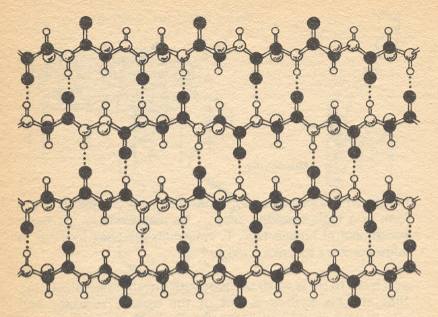
Figure above :
Another structure formed by polypeptides. Instead of coiling into helices, in the beta sheet the zig-zag chains of amino acids run side by side, held together by hydrogen bonds. The result is a soft strand - silk is made up like this.
(After GRIBBIN, 1987)
Armed with this kind of information, biochemists can now account for most of the structures in your body, the fibrous proteins which form its physical bulk. Collagen, for example, is the most common protein in the body. It is literally what holds you together, an important part of skin, tendons, cartilage, and bone. And collagen, like hair, is made of three-stranded helical chains, different from the triple alpha-helices of hair but with a family resemblance.
[In terms of Unimol, we may, I think, see the fibrous proteins as making up, together with other "molecules", the one living molecule. On the other hand, the globular proteins, especially the enzymes, seem to be part of the aqueous serum-like medium supporting the living molecule.]
The first X-ray diffraction pictures of fibrous proteins and cellulose date from 1918. Astbury's pictures with their tantalizing hints of regularity in the structure were obtained in the late 1920s and widely published in the early 1930s. Crystals of globular proteins were analyzed in the same way for the first time in 1934. But it took two decades for molecular biologists to begin to come up with structures to account for the X-ray diffraction patterns of even the simpler globular proteins. The story is one that I [Gribbin] only have space to touch on lightly here, but it is of the greatest importance in understanding how the body works. Indeed, the study of biochemistry is, in large measure, the study of enzymes -- and enzymes are globular proteins.
Enzymes are the molecules in the body that encourage, or inhibit, chemical reactions between other molecules. In chemical terminology, they act as catalysts, altering the rate at which chemical reactions occur [Without catalysts, most biochemical reactions are very slow, indeed, too slow, at body temperatures], without being altered themselves by those reactions. The best way to understand how this can happen is to think of a large, roughly spherical molecule -- a globular protein -- which has an indentation on its surface perfectly shaped to hold two specific and much smaller molecules. When those two different molecules sit in the cavity so conveniently provided by the enzyme, they will be held in alignment in just such a way that bonds can easily form between them. Instead of two molecules, we have one, and the enzyme can now release it to go about its biochemical business inside the cell, taking in two more small molecules (exactly of the same species as the first two) from the brew of chemicals around it and repeating the task as necessary. In a similar way, some enzymes break other molecules apart.
This is a gross oversimplification, but the image will do for our present purposes. Think of the enzymes, all of them globular proteins, as single-minded robots, each with its own specific task. One enzyme will join together one pair of molecules, a link in a polypeptide chain, perhaps, or [join together] the molecules involved in providing energy for your muscles. Another enzyme will be devoted solely to breaking one particular bond between one pair of organic molecules [Here, in this last sentence, we clearly see how one, perhaps unconsciously, attempts to let the concept of "molecule" to refer to the smallest possible compounds, i.e. to the simplest chemical compounds. Indeed, the last mentioned "pair of organic molecules" is in fact one single organic molecule.]. In many ways, enzymes resemble the idiot machine tools of a factory production line. How they go about their work is, as I have already indicated, a story that has occupied many textbooks, and, indeed, whole university degree courses. Let's just accept that they do work, and that their work depends very much on their structure. The relevant, and interesting, question for the study of the double helix [the DNA] is how they are themselves manufactured, and what [immediately] determines the unique structure of each globular protein.
[In all this about proteins, fibrous as well as globular, as they occur in the living body, we discover that each one of them has a structure that precisely corresponds to a particular function, i.e. corresponds to the ability, or makes possible, to actually perform a particular task for the benefit of the larger whole to which it belongs. Indeed, for a protein, the ability to perform a particular function most often depends on the 3-dimensional morphological structure and shape of the protein molecule, the morphological structure and shape rendering the molecule either to be a structural building block, or to be a biochemical tool (an enzyme or a hormone). But this morphological structure and shape is totally determined by the nature, number, and consecutive order of the amino acid residues making up the protein, i.e. its polypeptide chain. Even when, in making up certain certain proteins, in addition to the polypeptide chain there is often yet another important group, not of the nature of an amino acid, the very shape of the molecule is crucial for its function, and, as has been said, this shape and morphological structure is entirely determined by its amino acid sequence. Well, even relatively small proteins (like for instance myoglobin) may contain, in each of their molecules, several thousands of atoms, arranged in a single polypeptide chain made up from some 150 amino acid residues. Further, we know that in the world of organisms there are in all 20 different amino acids (see Figure above), as residues building up natural proteins. If we now imagine how many different strings, each consisting of 150 amino acid residues (in the case of still simple proteins!), are in principle possible, we come out with the horrifying number 20150, that is 20 to the 150st power. Even the whole known Universe cannot contain 20150 amino acid residue particles! From this multitude of possible proteins, having a length of 150 amino acids, a few, or even a single one is eventually "selected", one that can, thanks to its particular amino acid sequence, perform a particular biochemical task or one that can be a morphological building block of some organism or of one of its tissues.
It should further be noted that the structure of each polypeptide chain, wherever it occurs in a protein, is not given by some simple chemical rule, such as "the amino acid glycine is always next to the amino acid valine", or by simple repetitions, such as six leucines folowed by four valines and two cysteines, then repeat the whole pattern to the end of the chain. It really is best described in terms of a coded message. It is a sequence that has no rule, and yet it contains information. And to explain the presence of all that information in the protein you need a code. Every protein molecule, in other words, contains a coded message which ensures that each protein (especially the globular proteins) has a specific shape which uniquely fits it to carry out its role as a molecule of life. Where in the living cell is the essential blueprint which tells the cell how, and when, to manufacture each kind of protein? And how do the cell's engineers -- its own enzymes -- carry out the task of translating that code into messages in the form of amino acid sequences, proteins, such as insuline, hemoglobine, and the rest? We now know that this code lies in the double helix of DNA (desoxyribonucleid acid), the life molecule itself (DNA has been discovered already in 1869 by Miescher). So the problem of how a particular amino acid sequence is, from the astronomic number of possible sequences, selected to be formed, - an amino acid sequence that is functional, which here means that it can perform a particular task for the benefit of the whole (organism), - that problem, is now shifted to the origin of its code residing in the DNA molecule, here in the form of a sequence of nucleotides (constituents of the DNA), a sequence precisely corresponding with the amino acid sequence in proteins. And thus it is essentially the same problem : How can, from the astronomical number of possible nucleotid sequences, a particular sequence be choosen that corresponds to, i.e. codes for, an amino acid sequence representing a protein that can perform a particular function? And we believe that the model of natural selection cannot solve this problem. Chemical bonds form between atoms and atomic groups, - and thus molecules are formed, not as a result of being guided by some necessary functional end-result, but exclusively by lowest-energy configurations. In fact this lowest-energy configuration of atoms and atomic groups in molecules is the very criterium of "selection", not the nature of being functional of the molecule [being such criterium]. And of course there cannot initially exist a pool of all possible different protein molecules, different sequences of amino acids or nucleotides, even if all of them were energetically stable, from which [pool] certain molecules could be selected, because the possible number of 'candidates' in such a pool is much too large. So there must be agents that directly produce these special protein molecules, respectively, these special amino acid sequences or nucleotide sequences. But this can only be accomplished by some sort of final causality, and we know that in the material world there is no place for such causality. All causality in this world is efficient causality driven by lowest-energy configurations. Even the "final causality" in humans will, according to us, eventually turn out to be not genuine final causality at all, after the "mechanism" of the brain is fully, or at least further, clarified. Perhaps the phenomena of the human mind point to the existence of a purely "noëtic" domain of Being, an immaterial domain of pure "thinking". And indeed, Bohms idea of the Implicate Order, may fit in all this. And that's why we have, in the previous and present Part of Website (Part V and Part VI), worked out this idea further, especially to explain and interpret "organic evolution". We have found out that truly functional material structures, as we see them all over the organismic world, cannot be generated in the material world -- the Explicate Order -- all by itself. They have, as strategies, developed in the enfolded, immaterial world, the Implicate Order, in which the overall driving force for development resides in the strife or wish of immaterial forms to become material. And for this they must be able to exist and persist in the Explicate Order. In the Unimol view of organisms, it is the single living molecule that wants to exist (in the Explicate Order) by being itself a strategy-to-so-exist. This strategy consists in the fact that the living molecule is constantly supported by a non-living medium in which it is embedded, and which is the molecule's most direct existential condition. Living molecule + support medium together forms the strategy in a broader sense, in fact the strategy for the molecule-and-its medium to exist. And this broader strategy -- the organism -- is itself in turn embedded in its proper environment, its ecological niche. While the mentioned support medium of the molecule does form with this molecule one material (though not chemical) whole, the organism, - the latter does not form such a whole with its ecological niche.
So, according to our "noëtic theory of organic evolution" the functional proteins, and their corresponding codes in the form of nucleotide sequences in the DNA molecule, have not gradually evolved from initially simple non-functional proteins/nucleotide sequences, but directly derived in the Implicate Order and being projected into the Explicate Order in the form of organismic species, replacing other species in some particular environment.]
Having now concluded the above few sections mainly taken from Gribbin, In Search of the Double Helix, 1987, sections concerning amino acids and proteins, and us having commented on things, we now shall pick up Oskar Müller's text again, the text on The world of the renounced dimensions, investigating the possibility of very large organic molecules, molecules having the size of whole organisms, that is, we continue with the Unimol view of organisms.
Especially stable bridges across -S-S- must be possible between two cysteine groups [in a polypeptide chain or between two adjacent polypeptide chains], "so that a cystine molecule simultaneously belongs to two different protein giant molecules" [i.e. one single cystine molecule belonging to two cysteine molecules (See Figure above)]. But aren't two giant molecules firmly [chemically] connected with each other not the same as one single even larger giant molecule? But even if one concedes this, one seems to want to draw some practical limit, a limit thus, when "some" chains are side-chains and so connected chemically again. Why one refuses to see many and then finally all connected among one another and chemically dovetailed? Why does one speak of group systems [each of them] consisting of a still surveyable number of atoms, and at most adhering to one another, and at best grading into one another (what in these dimensions only makes sense in the form of chemical bonding)? [Müller here means that often a given large molecular complex is seen as a system of groups, i.e. as molecules united (in groups, and then in a system of groups). So the complex is not seen as a molecule of its own, as it should be].
The continuously recurring intention : to "demarcate", within larger wholes, the continuity of "protein molecules" (after the known fragments) [i.e. the intention to distinguish within a larger whole the "true protein molecules", seen as mere fragments in another view, and so to interrupt the true continuity between these known fragments], and to still see them as such [as protein molecules] [i.e. continuing to view the delimited parts as "protein molecules"], has in fact no parallel in the case of small molecules, because there one is not explicitly taking care of not to forget the continuity between the constituents [of such a molecule], namely the continuity between the atoms and atomic groups.
Looking at some one isolated and still definite macro-molecule, one is of the illustrative opinion that every atom of it, as a result of the chemical overall bonding, belongs in one way or another to all remaining atoms of this molecule. If one connects two such equal or different macro-molecules by a real, chemically fully capable of carrying load [without breaking] (not to confuse with : preferredly breakable) bond, for example over multi-valenced atoms, then one focusses upon seeing, purely graphically, the new connection as a "bridge", and creates a gross relational-in-essence-difference between atoms belonging to the same previous partner, and atoms belonging to other previous partners, meaning that one ignores the full validity of the bridging bond, and does not recognize that the generated double-molecule is a new unrestricted single-molecule. With this, one often enough is right in practice (and, in the case of many weak bonds -- especially often in H-bridges ( NOTE 344 ), which we above didn't include anyway -- [is right] also in the "theory"). It is, however, evident that there really and certainly logically are countless cases in which the bridging creates a new undoubtable full-molecule with equivalent indistinguishable unity of the relationship-in-essence between the components, in such a way as if one had connected two "half " cellulose molecules into a complete one, or if one had connected two peptides with each other by the peptide bond.
It is -- taken into account the far-reaching possibilities one has to do with -- a peculiar and badly founded attitude not to recognize in the "net-like" connection, or, a connection, realized through "local joints" ( NOTE 345 ) (of which one doesn't deny at all that this connection is a chemical one) of several or many definite molecules, the origin or formation of just as real a macro-molecule.
That within this macro-molecule may be created complex new (!) structures such that one cannot speak anymore of a delimitation of definite sub-molecules [as it still can in di-, tri-, poly-, etc. forms], is an elementary fact for the chemist, for he is used to see diphenyl just still as a benzene, but not so anymore does he see a complex indanthrene dye. [The "phenyl group", -C6H5, is a benzene-ring of six carbon atoms. Five of these carbon atoms carry a hydrogen atom, while the sixth connects the group to some other group.] Here, as in the case of the organismic macro-molecule, the internal delimitation, at most asked-for out of analytic or synthetic interest, is difficult, but not so the essential external delimitation [where the molecule, as molecule, begins, and where it as such ends]. The organismic macro-molecule doesn't lack the posibility for it to be defined sufficiently "rigid" and to be determined, if one, avoiding naive analogues with the micro-molecules, takes into account the proper nature of the organismic ( NOTE 346 ).
Today one is inclined to take as macro-molecules : molecules of up to 10 - 4 cm. That is four powers of ten above the atomic order of size. Not taking into account questionable considerations concerning stability and limiting sizes, we should like to say that the chief difficulty as to stability has been, in the mentioned order of magnitude, overcome anyway, and that there are no fundamental objections to increase the order of magnitude with 5 or 6 further powers of ten, insofar as the generated new forms functionally make sense at all, which [unlimited increase in size] is excluded for inorganic forms (except for certain, but very vague, limiting cases of giant crystals), but demonstrably favorable for organisms. And in order to realize such a moment of favorability we may well entrust to Nature the necessary conditions [for size increase].
A self-limitation of magnitude, residing in the structural principle of construction, can only be seen in inorganic forms or things, and thus, on the one hand, [seen] in the atoms with their stability limit at uranium (the natural trans-uraniums are merely a variation) [If we go up from the lightest atom, hydrogen, through heavier atoms such as sodium, iron, etc. (where "lighter" more or less implies "smaller", and "heavier" more or less "larger"), we finally arrive at the heaviest atoms, such as uranium, and there we meet for the first time (internal, intrinsic) instability of atoms, expressed by their radioactivity.], and [seen] in the [inorganic] molecules on the other [hand] with their size limit as a consequence of external (extrinsic) stability. Only the living molecules breach this limit and namely in a way which one might characterize as not limited at all anymore, because the practical organismic size limit is a favorable functionally morphological one, not a theoretical [i.e. the size limit in organisms is biologically determined, not physically].
It cannot be missed that in the above mentioned short selection from common views there are certain clues for the organismically macro-molecular. In fact, we see them everywhere, but they also are broken off untimely with geat unanimity ( NOTE 347 ).
As "fruits" of lengthy auxiliary images [hypotheses] once one had [as it seemed] succeeded to "explain" the movements and changes of shape in organisms by means of physically chemical phenomena such as osmotic pressure differences, hydratation and dehydratation (with swelling and unswelling) etc. in and on a colloid-chemical system, until one surmised and subsequently also found confirmed that it is the fibrous protein substance itself which reversibly elastically contracts after stimulation ( NOTE 348 ). Today nobody will doubt anymore that, for example, the acto-myosine "particles" have a molecular structure through bonds, and that the contractility is an intra-molecular structural or super structural change under functional constancy of the bond total.
Relating contractility with protein gels is nothing else than the attempt to establish for an unknown domain the connection with a known one, here by means of the utilization of a colloid-chemical concept. We do not want to say that this is fundamentally wrong (for the vital protein, living in its medium is, among others, also an internal gel), but one also doesn't gain very much by so viewing things. If one approaches these things from another angle, namely from the Unimol constitutional view, then one already has an all-embracing explanation and may grant the incidental, such as, for example, gel-behavior, a changing, but always subordinated, significance. Unimol relieves from all those elements, which outside Unimol become cumbersome and often enough become empty vehicles of significance-out-of embarrassment.
What in the beginnings of chemistry, before real atoms and valences and the like were known, has led to the characterization of pure substances (physical constants, uniform look, uniform properties, and others) must now once again be repeated at the level of the organismic. If one then, with this extended and amended knowledge, again engages oneself in the analysis of Life -- which now is not anymore pure elementary analysis, as learned in inorganic chemistry -- then one would, following the same approach as was followed in the early days of chemistry (note, that any method always is reason-borne speculatively hypothetical), find the true building blocks and the true elements-to-combine, constituting Life.
The sharpest and definitive means of proof for Unimol do not consist in uncovering the continuous chain of [chemical] bonding, for simply optically this already isn't possible at all, and the most conspicuous aspect of bonding, namely its "firmness", fails to materialize because already freezing water can break it up. The next conspicuous aspect, namely the (physical) properties, also fails [to prove Unimol] because two organismic supermolecules can hardly be distinguished, practically, qualitatively, from their true association-through-bonds [i.e. a giant molecule doesn't acquire new general physical properties just as a result of the fact that it is an association of two lesser giant molecules, i.e. doesn't acquire physical properties that only express this association. (Of course it may acquire new chemical properties, because the association has resulted in a different molecule.)]. Unimol may only be demonstrated as to be consequent and unavoidable by setting up a suitable theory of bonding and by thoroughly clearing up the biologicallly oriented atomic-potential framework.
We do not want to exactly pin us down on the nature of all bondings [in a living molecule]. A substantial part of them must be very similar to the homopolar [covalent] bond of protein equivalents. Generally seen, it is [in Unimol] not about a complete conformity of all bonds with those how we describe them in protein equivalents with the atomic and molecular formula language. This is already ruled out by the special state [of the living molecule] demanding and implying deviating bonding types. Among them, such deviating types may merely be similar to ground and limiting states. But in addition also very special and strongly deviating bonding types may appear, not similar to the homopolar bond anymore. Only one condition must be satisfied : The types of bonding, generally taken, must be [intrinsically] defined or definable and result in a true connection, a connection which places "the whole" alongside the inorganic molecules, and especially also alongside the organic classical micro- or normal molecules, as equivalently molecularly ordered. Under the necessary and legitimate -- and not essentially altering things -- broadening of the concept of "molecule", organisms remain precisely that what they were since the origin of Life ( NOTE 349 ), namely genuine molecules, physically chemically meaningfully defined -- which is not equal to precisely as exact a describability as, for instance, an urea or lecitine molecule -- and demonstrated to be true uni-substances on the basis of all of the unchangeable and sharply delimited properties of their own.
As most significant fact we therefore must stress that in the organismic realm [organisms] it is not about a combinatory system of, it is true, relatively large and isolated individual molecules, that is macro-molecules still being small as compared to the size of an organism or a cell, but about each organism to be one single organismic molecule. In it, there is no end of bonding [i.e. the organismic molecule has, as molecule, not reached some intrinsic size limit], but one always has seen things as if there were one. It is peculiar that this end is not doubted by anyone, respectively stepped over. This would be reasonable if assuming no end of bonding in an organism would have absurd consequences, but one can, if need be, call these consequences enormous, but, indeed, "enormous" in the positive cognitive sense ( NOTE 350 ). It is even very difficult to illustrate this end, this limit of bonding extension, because every simple table of sizes (orders of magnitudes) fails, in the relevant domain, to show any break or discontinuity, and shows a regularly increasing series [of magnitudes], where the classical macro-molecules are followed by the virusses, bacteriophages, vaccins, and micro-cocci. Certainly, we do not want to bring in this series of increasing magnitudes as (physically chemical) proof of Unimol, but want to establish that there is certainly no discontinuity here.
Unimol brings with it the fact that, as a result of the enormous size and extension of the molecules representing it, the concept of "molecule" acquires an extension of meaning [an increase of content] hardly conceivable in the realm of small molecules, for there the concept of molecule can be characterized with 5-6 features, and vice versa. Out of reasonable grounds and for the sake of understanding, we ask not to see in Unimol a problematic over-extension of the concept of molecule ( NOTE 351 ), but rather not to see the molecule concept of inorganic and organic chemistry [ordinary chemistry] as absolute and unchangeable point of departure, but to see it there as [the result of] a reduction and simplification of the molecule concept to the most simple, and there [in ordinary chemistry] sufficient, form. But, with this, of course, not embracing the other extreme by viewing the inorganic molecules as lower or even imperfect individualities [imperfect individual unities]. The usefulness of the proposed view [Unimol] is evident, there is no decisive agument against it.
The energetic behavior of living substance often is seen as a feature especially characterizing Life, namely first as to the apparent desentropic structure and action ( NOTE 626 ), and secondly as to the energy storing and the ability of sudden release. The first only appears in a superficial and partial consideration [meaning that in all (macroscopic) processes entropy is produced (such that the total amount of entropy in the Universe has increased)], the second is evident and specific. Insofar it is about gross-energetic, that is, generally about macrophysical processes, such as muscle work, it is outside our special interest. Precisely that what interests us is an aspect that can be approached from two sides :
The energy-need [of an organism] perhaps boils down to maintain in a reception process [a buffer process] an intermediate in-statu-nascendi [= in a state of becoming] state having once appeared as a result of the origin of Life and subsequently nucleotidic-uteroidly [i.e. in reproduction] ever repeating ( This maintenance of the in-statu-nascendi intermediary state being like keeping a wound open), and to buffer against the threatening denaturation process. The interconnection of the processes necessary for it is the "system" of life, the enduring [internal] connection of the living being is the first perfectly formed multiply performing living being, the cellular aggregate.
What image may one form of the energetic "charging" or "tension[ing] of living substance", that is, [the energetic charging] of such structures that all in all are stable as to their atoms, and only vary in a not even essential part of their configuration -- to which part one can assign an energetic equivalent. As to the energetic tension, we do not refer to the metabolic processes, which most often proceed through coupled reaction cycles, but only to the, certainly small, energetic equivalent that must be supplied to the living substance in order for it to be able to maintain its special state as such anyhow. So here it is not about the qua amount extraordinary energy balance "sheet" at the living substrate, but about the small but important energy balance sheet in the living substrate.
[I shall try to explain the foregoing as best I can : This living substance is one particle amidst many other particles - the one living molecule in its multi-molecular support medium. As such it is a system that can be discussed from the thermodynamic viewpoint. And indeed, this is about making up the energy balance "sheet" at the living substrate and it is thermodynamically evaluating an inter-molecular system. If, on the other hand, we only consider the living molecule itself (and not its support medium), then we have in fact only one "particle" to consider thermodynamically. So in order to correctly consider this molecule thermodynamically, we shall view it as an intra-molecular system, a system, that is, consisting of virtual particles. And indeed, this is about making up the energy balance "sheet" in the living substrate.]
Where, especially, does it correspond with the classically normal? Well, no matter whether one is speaking about material energy transport (ATP) or with intra-molecular transfer (electron conductivity), in the organismic giga-molecule there surely are functional spots without any special mechanism, but with something that we as type do already know, albeit that we cannot directly say which of the already known it is.
Truly connected to the living molecule are undoubtedly places or regions that at their distal end have a "crystallogeneous" normal-configuration and there may experience periodically regenerative processes of conversion with positive energy balance shifting [from the distal] through the proximal end onto the living special-structure. This would, in accordance with our remarks in the Section "Denaturation" (previous document), mean that Life, also in its reduced form, "lives" not only in an inorganic environment, but still being truly connected with the non-living inorganic.
Such connected-to's, lying beyond that what one takes to be the true material basis of Life, the truly living substance, we unequivocally meet in the prosthetic groups [specialized chemical groups bonded to the polypeptide chain] of the enzymes. The mediating part is played by the apo-groups [= the protein portion of the enzyme], which too are "still" not living substance in themselves, but which may be [spatially] adjacent to it. In higher organismic life there are many such adjacencies, for instance bones, cartilage, horn, etc., which practically in a bonded fashion lie adjacent to living substance, without actually being such a substance (or apearing not to be such anymore).
These casual remarks will be sufficient, because it is about too little known a detail-phenomenon.
Much more substantial can be said from the standpoint of general thermodynamics and special entropy, although also here the opinions differ more or less. As being evident one should presuppose that the chief thermodynamic laws [1st : conservation of energy, 2nd : increase of entropy (ultimately in the Universe as a whole) as a result of any (macroscopic) physical or chemical process] must prevail unrestrictedly, meaning that genuine ectropy [in contrast to entropy] is merely a confusion and that all des-entropic effects (precisely as in the inorganic realm) are only to be found in partial systems [i.e. a natural system of interacting entities, but a system of which one part is considered while omitting the other part from consideration. In such a partial system often entropy seems to decrease instead of increase]. Finally one should note that the living substance, as to its structure (with or without Unimol) strongly deviating from the normal, does not offer any normal places [or opportunities] to act on for the thermodynamic rules and laws, and seeming deviations will occur if one doesn't take this into account.
So little is known as to what precisely the living protoplasmatic state really is, that one even cannot definitively say that thermodynamically the living substance is an isothermous system in which the transition from free energy into heat is irreversible. This then is not important when the living substance takes care that that fraction of the free energy that becomes heat is held at an appropriately low level or at most being functionally exploited for upholding the warm-blooded state among other things. ( Living substance preferably "operates" with chemical intra-molecular energy involved in exchange.)
Even when one considers unimol as individual substance in itself - and thus removes it from its context, which is absolutely illegitimate -- one will say that the organismic molecule is too big for it to be possible to non-statistically exploit the Second Law of thermodynamics [the Law of ever increasing entropy] which [law] is a probability statement very much accessible for [local] deviations. The possible deviations may be the same (not essentially more, nor less) as they occur with and on micro-molecules, and in the present case on quasi-micromolecular "ends" of the macro-molecule ( NOTE 630 ). However, one cannot completely exclude that nevertheless there do exist certain possibilities of deviation in the [single] macro-molecule, because inter-molecular statistical uniform distribution this time takes place intra-molecularly [and thus the non-statistical having beome statistical].
The participation of the organismic macro-molecule in chemical reactions appears to recomment to combine the mathematically thermodynamic treatment (referring to the macrophysically-taken overall happening) with the especially molecular-kinetic consideration, in order to keep down difficulties and contradictions by means of elements adapted to reality instead of questionable auxiliary hypotheses.
In the next paragraphs Müller thermodynamically discusses the living macro-molecule, not so much as belonging to a system of many other molecules -- the micromolecules [and the macro-molecular nucleic acid molecules] of the support medium -- a system as a statistical collection, but [discusses this one living molecule] as a "system" of the parts of that one molecule, as already indicated above. In so doing, it is possible to thermodynamically discuss one single molecule, the living molecule. In fact the parts of this molecule are chemical entities in interaction, and therefore, in a way, the thermodynamics of chemical reactions can be applied to just this single molecule. And for all chemical and physical reactions whatsoever the expression dH - TdS = dG is fundamentally important, important for understanding not only inter-molecular processes or interactions, but also the intra-molecular interactions in the one large molecule. For a good, at least qualitative, understanding of this thermodynamic expression we will quote and paraphrase a section from Introductory Chemistry written by Ouellette in 1970 :
There are many physical and chemical processes that occur naturally. The flow of water downhill and the expansion of a gas to fill a container are two examples of spontaneous physical processes. The reaction of hydrogen and oxygen to yield water and the burning of wood are examples of chemical processes. Analysis of all naturally occurring processes reveals that two fundamental features control them. Most systems tend to produce a state of lower energy and as a consequence release the net energy difference between the initial state and the final state. The reverse process is less common and can occur only if the second controlling feature, entropy change, is favorable. The flow of water downhill and liberation of heat in the formation of water from hydrogen and oxygen occur because a state of lower energy is achieved. In such cases the energy difference between initial and final states is called the change in enthalpy, dH. By convention the liberation of energy is given a negative sign. The second controlling feature of all processes is the tendency of a system to achieve the most random or disordered arrangement [of its constituents] possible. This feature is important in the expansion of a gas or the vaporization of a liquid. The degree of randomness or disorder is called the entropy, S, of a system. By definition the entropy change, dS, is positive for increasing disorder [and negative for increasing order]. The entropy change counterbalances unfavorable enthalpy changes in some systems. If the increase in the degree of disorder is great, an endothermic process can occur [In exothermic reactions energy is released to the system's surrroundings and thereby preserving the order in the system because entropy is given off to the environment. In endothermic reactions, on the other hand, energy is taken up by the system from its surroundings increasing the energy of the system, but if this is more than compensated for by the increase of disorder of the system, the reaction still proceeds spontaneously.]
The temperature is a significant factor in determining the relative importance of enthalpy and entropy contributions to a system that can undergo change. At absolute zero, all substances are [internally] ordered and entropy differences between two or more substances are zero. Therefore, only their relative energies determine their relative stabilities. With an increase in temperature a variety of molecular motions become possible, and the tendency toward disorder changes from substance to substance. The difference in entropy can play a variable role as a function of temperature. At extremely high temperatures the entropy differences between various substances may play a dominant role in the course of a reaction.
The relationship between enthalpy changes and entropy changes is given by the following expression, in which dG symbolizes the change in free energy of a system at constant pressure :
dH - TdS = dG The free energy change, dG, is a measure of the driving force of a reaction or the tendency of it to proceed spontaneously. When dG is negative a chemical or physical process occurs. The negative enthalpy change, dH, [this when energy is released] previously described as being important in determining the course of a reaction can be seen to contribute toward making dG more negative. Similarly, a positive entropy change, dS, [and thus a positive TdS term] contributes toward making dG negative. From the expression it can be seen that dH is more important at low temperatures and dS becomes more important at high temperatures.
The power of thermodynamics stems from the potential to be able to calculate whether or not a hypothetical reaction will proceed in a desired manner. If, after calculation, dG is negative the reaction can proceed spontaneously in the desired direction. However, the difference in free energy does not indicate the velocity of the reaction since the rate is dependent on the activation energy [the energetic hurdle to overcome for the reaction to start off ] and not the difference in energy between reactants and products. Therefore, the spontaneous or naturally occurring processes may proceed at very slow rates.
The size of the free energy change, dG, between reactants and products determines the position of equilibrium. For a reaction with negative dG the equilibrium constant is larger than 1 and the reaction goes to the right. When dG is positive the equilibrium constant is less than 1 [and the reaction goes to the left].
* * *
In this, yet another thing appears to us remarkable : While one already often has the impression as if the organism worked des-entropically, then, on the other hand, one may likewise surmise that it often steers the for us visible or assumed processes such that in the relation dG = dH - TdS ( NOTE 631 ) the entropy term TdS, in spite of the relative low temperatures [in all organisms], becomes rather large, precisely then when dH would [otherwise] disturb things [i.e. when dH is positive, then, as a result, dG (free energy difference) will be positive and the reaction not proceeding spontaneously. But if the TdS term is high enough, dG will become negative again and the reaction will start off spontaneously]. Indeed, the body can, apart from the "inorganic" processes (phosphorilation, and others), rarely do something with free energy, and must attempt to get rid of it or functionally exploit it. The versatile and complex organismic substance is well adapted to hold quite high the, macrophysically thermodynamically not exploitable anymore, amount of energy dissipation [the TdS term in dH - TdS = dG ] [i.e. to hold the export of entropy at high values] and, in the microphysically chemical sense, yet to exploit it still further. For it is the dissipation amount distributed over the single inner molecule.
In this pseudo-mechanism, ultimately realized by the fact that a recognizable chemical type-reaction energetically (i.e. only by exploiting the with it occurring energy dissipation) is continued with an uncountable number of subsequent "small" chemical processes of intra-molecular nature (the otherwise considered states of affairs always refer to inter-molecular processes) ( NOTE 632 ), [in this pseudo-mechanism] we meet an essential minimal-partitioning element of the organism. The physical forms which, thanks to a very large number of degrees of freedom, have taken up the having-kept-very-high entropic dissipative energy and possessing it formally only as merely a retention of simple "inexploitable" heat, are in the organismic [i.e. in the organism] at the same time chemical entities, whose constitutive particular nature may allow to have things followed by yet uncountable single- and intra-molecular transformations, making up in persona a large part of the life-characteristic functional [aspect of the organism]. With all this, in the overall system the physical notion of entropy is in no way compromised neither to the one nor to the other side.
[Here we have, following Müller, spoken about "intra-molecular chemical reactions", i.e. reactions inside a single molecule. One might wonder how this is possible. Well, it is possible in the following fashion : The single giant living molecule is an utterly complex protein-like substance. It is a dense tangle of billions of atoms connected by various chemical bonds. As such it is a flexible labyrint of interconnected chains and side-chains everywhere in contact with the support medium (via denaturation membranes). Parts of particular chains may get connected with parts of the same or other chains of the molecule. This can be done by hydrogen bonds, but especially by the stronger sulfur bridges. Indeed, when a peptide chain (as part of the organismic molecule) is folded in hair-pin fashion, two cysteine residues (as two of the many different amino acid residues making up the polypeptide chain) may come close to each other and may then, under suitable conditions, be connected by a sulfur bridge as was already shown ABOVE. This, of course may also happen between two different polypeptide chains (of still the same organismic molecule). And, of course, also more than one such bridges may form at the same time. And in different conditions (as a result of local changes in the support medium) such sulfur bridges may get undone again. So here we have an example of chemical reactions within a single molecule. And it is now evident that the organismic molecule is far from static, it is highly dynamic. And while in the inorganic domain a given molecule becomes metaphysically a different Substance (and also, of course, chemically so) as soon as its chemical constitution is altered, we must suppose that in truly organismic molecules, living molecules, this is not so. Such a living molecule, when chemically altered, becomes, it is true, chemically a different substance, but metaphysically it remains the same Substance, the same "self". Why this difference, this apparent inconsistency? Well, while in the transition from one non-living molecule A into another, B, the latter certainly must have lower energy (dG is negative), and in that respect A is ordered to B. And this holds for all chemical reactions whether inorganic, organic, or organismic. But, when energetic conditions are satisfied, A may also change into C, or D, or whatever other chemical substance. In the case of the organismic molecule, on the other hand, things are different. Here, apart from cleary accidental chemical change (often noxious), a given chemical substance A is ordered to eventually become substance B (and B is in turn ordered to eventually become C, etc.). And it is this "ordered to" that guarantees that metaphysically the living molecule remains what it was. We here, of course, are thinking of the individual development of a given organism, including complete metamorphosis such as we see it in many insects. In all this, the "self" of the living molecule remains the same. All the changes mentioned form part of the strategy of the metaphysical material Substance to exist and persist.]
This [proposed intra-molecular thermodynamic system, in the case of an organism] is just one, and, moreover, for the time being a pure construct of reason. As a result of changes in concentration, [and as a result of the presence of ] acceleration- and deceleration-substances and many more, additional regulations may be realized. But our above proposed regulative mechanism is especially simple and typically adapted and may be capable of replacing many considered complex and in fact not quite understandable and very lengthy system-mechanisms. We, anyway, believe that as a result of the uni-molecular "dynamics" with its intra-molecularly independent enfolded functional equilibria, only a minute fraction of that cost is needed that is demanded by the so-called dynamical, but comparatively static systems of interaction with their obscurely many adjacent substance-equilibria. In the "system" [i.e. as conceived in the system-approach, versus Unimol approach] all goes into the "infinite", because : the more complex, the more vulnerable to disturbances (perturbations), everyone of which [disturbances] has to be compensated for by a new mostly more-sided mechanism of regulation.
With this we conclude our survey, but not without considering a condition closely connected with our problem (of energy household in organisms), namely structural conditioned energy transport. [Although the subsection - in Müller's text -- on energy transport is interesting, it has only relatively little bearing on Unimol. So, at least for the time being, we skip it and proceed with the next Section.]
The by us rejected pure system view of the organism, that is, the view of the ordered harmony of individual micro- [or meso-] molecular components, in fact has a very peculiar tale of origin, which -- as to the demand of scientific credibility -- dwarfs the [theory of the] origin of Life.
If one wants to relate Life with chemical compounds at all, and if one is put off by the assumption that some small primordial slimy lump suddenly began to crawl away (and moreover slowly having changed into an elephant), then one must admit that in the course of origin and development of Life, as well as in today's cellular life, genuine uni-molecular stages appear and are passed through. On the other hand, in the system-approach one believes that genuine life originates as a result of such molecular forms mutually engaging in specific relationships ( NOTE 850 ), and thus forming a multi-component system, which, as a whole, lives, and for whose wonderful development natural selection is rightly held to be responsible.
In all this, it is important, and in most cases overlooked by the supporters of the system-approach, that it is not only a question of cooperation, but especially also of interaction between the assumed uni-molecular components. Consequently, there must exist among them average relationships, relationships that are robust, constant and fixed, and thus there must exist specific orientations having their point of departure in one, and their effect in the other molecular partner.
After rightly having taken our body in its totality to be a chemically physically material system with an enormous number of definable, and thus molecular substances, meaning that in our body there is a huge number of individual molecules present, one may ask :
The free, but not isolated in infinite dilution, molecule is carrier of a determined "field of force", whose nature and magnitude depends upon its structure. Accordingly, in interactions with its fellow molecules or with other molecules relationships do appear that only in the not-realized ideal case are wholly pure mechanical material interactions, otherwise, however, become essentially modified as a result of the inter-molecular forces which are residues of effects of constituents.
The most general description boils down to the fact that between molecules do exist, with increasing mid-point distance, proportionally rapidly declining repulsive forces and slowly declining forces of attraction ( NOTE 851 ). The latter constitute that what one usually indicates as inter-molecular forces, and less well indicates as Van der Waals forces [Van der Waals forces are (weak) attractive forces between free molecules. They result when of a molecule A its electron cloud (carrying negative charge) is mostly, as a result of deformation, concentrated at, say, the left-hand side of the molecule, such that one or more atomic nuclei, which are positive, are more or less exposed at the right-hand side of that molecule (only shielded by a thin screen of electrons), while the electron cloud of a molecule B, being, at that moment, at the right of molecule A, is mostly concentrated also at the left-hand side of the molecule, resulting in the fact this molecule is a bit negative at its left-hand side. And now the result is that the molecules A and B weakly attract each other electrostatically.] ( NOTE 852 ), and the effective real material situation constitutes itself from the superpositioning interaction of the order-creating inter-molecular forces on the one hand, and the disorder-creating thermal motion on the other. And because the latter too strongly intervenes into the former, and only, if weaker, degenerates the interaction system into the counter-effective and counter-functional ossification of the crystalline condition, one cannot, in the "simple" inter-molecular forces, see any means by which they would produce very special metabolic but functionally stable qualitative effects of ordering ( NOTE 853 ). Such effects of ordering should outstrip the merely swarm formation resulting from unstoichiometric polarizational association, and the same goes for the formation of quasi-stoichiometric "super-molecules" [also these should be outstripped].
For the stability of super-molecules, -- as "kinetic" units [consisting] of basic molecules saturated as to chief valences, basic molecules, that is, loosely bonded by inter-molecular forces of di- and multi-polar nature as the sole collective action principle, and showing an equilibrium obeying the mass action law, -- is much too bad for organismic, exactly unequivocally continuously and reliably functioning, systems ( NOTE 854 ).
Simple systems with only one or a few species of molecules dispersed in a homogeneous medium, superficially do indeed give us the image of a stationarily unequivocal constant, a little varying with temperature and concentration, state. But even when the life span of the aggregational super-molecules [temporarily formed from the constituent molecules] is very long as compared with the time span between thermal jolts, then, in a complex multi-component system we cannot expect any other order than the microphysically ceaseless trans- and dis-ordering [formation and disintegration of order], which only macrophysically -- statistically summing up -- shows a statistical non-individual number- and material order [meaning, that only on average one may see a pattern or order]. But Life needs, at least largely so, the individual temporal identity, conformity, and continuity, and these are only realizable if the whole is a single true individual, the aggregation mechanism of which must therefore be the most robust one there is, and that is the true chemical bonding through so-called chief valences and not through one or another residual bonding force, the true chemical bonding, that is, which is resistant to the normal-physical perturbations. The coupling relation between cooperating system molecules must be such that it coincides with a true chemical bonding. [An example of bonding through a residual bonding force might be the formation of OH3+ ions from H2O molecules and hydrogen ions (H+) through the so-called dative bond [not to be confused with the hydrogen bond] by means of electrons not involved in covalent bonding. The valence of oxygen (O) is 2 and of hydrogen 1, so a normal compound between the two (through chief valences) is H2O, i.e. OH2 and not OH3 ]
At the same result as having just arrived at through a demanded postulate, one also will arrive from the practical side, where one lets assume the forces of aggregation the stability of the chemical bond ( NOTE 855 ), which can be realized in no other way than letting these connections to become true chemical bonds ( NOTE 856 ).
So : Because the "normal" inter-molecular forces, active between the relatively small protein molecules, decline with high [higher than square] power of distance, and, moreover, the distances incessantly change as a result of thermal motions, one needs, in order to have close interactions ( NOTE 857 ), a robust neighboring fixation which is only reliably fully-functionally guaranteed in the form of a true chemical bond, in which, in spite of this, the partners still enjoy significant system-freedom, which [freedom], in the case of very strong only-inter-molecular forces such as in crystallization (with the pre-stage of a strongly anisotropic liquid, or the pre-stage of a liquid crystal) and perhaps also as in physically-taken antibody/antigen aggregation, they [the partners], in large measure, lose [this freedom].
[Here we temporarily interrupt Müller's text on intra- and inter-molecular interactions, in order to explain its content still better. Especially it is how protein molecules behave when embedded in water as they are in living organisms. In all this, it is important to notice that water -- just liquid water -- is not just a random collection of individual water molecules, but in fact a network, a three-dimensional hydrogen-bonded jigsaw of water. Proteins embedded in liquid water may influence the structure of this jigsaw profoundly, gathering a layer of water molecules around them that are differently arranged than those far away from the protein molecule. And by this layer the protein molecule itself is in turn affected such that it folds up in a particular way. In the ensuing Intermezzo we must keep in mind that the mentioned authors (ELMORE, 1968, and BALL, 1999) cannot be supposed to adhere to the Unimol view advocated by MÜLLER (1959).]
Speaking, in Müller's text, about inter-molecular connections, especially of such connections between individual protein molecules, it is perhaps instructive to expound how they have their analogues in intra-molecular connections. In the latter connections we first of all think of true covalent bondings between groups which here are the amino acid residues connected to each other by the covalent peptide bond (which resonates between a double bond and single bond state and is therefore not able to twist about its axis). However, there may also be bonds, non-covalent bonds, between distant parts of the same protein molecule which bonds are responsible for the conformation of the molecule, i.e. responsible for the way such a molecule folds up resulting in a specific three-dimensional shape of the molecule which in turn is essential to its physiological function. Such non-covalent bonds are hydrogen bonds (see HERE (document "The Chemical Bond" in First Part of Website)), which also may play a role in inter-molecular bonding in proteins, and so-called hydrophobic bonds. As to these two non-covalent bonds in proteins it is instructive here to insert an exposition given by ELMORE, 1968, "Peptides and Proteins" :
A hydrogen bond can exist when two electronegative atoms, one of which is [covalently] bonded to hydrogen, can approach one another closely with the hydrogen atom preferably linearly placed between them. The hydrogen end of a bond between hydrogen and an electronegative atom carries a fractional positive charge and is attracted towards an unshared pair of electrons in another electronegative atom :
A--H ... :B If the two electronegative atoms involved in the formation of a hydrogen bond are identical [such as in ice], the hydrogen atom is shared equally between them. In any case, when hydrogen bonding occurs, the distance between the two electronegative atoms concerned is less than the sum of their van der Waals radii. Although hydrogen bonds have low bond energies of the order of 5 kcal/mole, there are so many groups capable of forming them in proteins that they play a large part in determining the molecular conformation.
Polypeptide chains tend to fold in such a way that the nonpolar side chains of amino acids such as leucine, isoleucine, valine, phenylalanine and tryptophan (see ABOVE ) tend to come together. The term "hydrophobic bond" has been coined to describe this type of noncovalent interaction, although it tells us nothing of its nature. Van der Waals forces between atoms are likely to be relatively unimportant [as to the formation and persistence of hydrophobic bonds] except when very close approach is possible, since the energy of bonding for this type of interaction [the hydrophobic bond] is inversely proportional to the sixth power of the distance between the atoms involved. Secondly, overlapping of pi-orbitals [I presume p-orbitals] may occur when a suitable steric relationship exists between amino acids with aromatic [i.e. involving rings of carbon atoms] side chains. This type of bonding is also likely to be of little consequence in determining the conformation of protein molecules, since many hydrophobic bonds involve amino acids with aliphatic [non-cyclic chains of carbon atoms] side chains [So in hydrophobic bonds in proteins overlapping of p-orbitals in most cases does not play a role, it is not a main source of hydrophobic bonding in proteins]. The most important source of hydrophobic bonding appears to stem from the interaction between apolar side chains of amino acids and water molecules [For the organization of H2O molecules in bulk ice or water, see next two Figures]. Water molecules in the vicinity of apolar groups possess a greater degree of organization than in the bulk of the solvent. The water molecules adjacent to the apolar structure have limited possibilities for forming hydrogen bonds with other water molecules and they therefore assume a regular arrangement in order to achieve the maximum degree of hydrogen bond formation. If two or more apolar groups can come into sufficient close contact, they exclude water molecules and form a kind of intra-molecular micelle. The number of water molecules which have [with each other] a highly organized structure near a cluster of apolar groups will be less than the number of water molecules which would be structurally organized around the several apolar groups if the latter were separated. In other words, the entropy of the solvent is increased if apolar groups come together and this is the thermodynamic driving force which favours the formation of hydrophobic bonds. It is not surprising that protein molecules tend to be folded in such a way that the maximum number of hydrophobic bonds are formed in the interior of the molecule, and amino acids with hydrophilic side chains tend to occur on the surface of the molecule.
Addition of organic solvents to aqueous solutions of proteins tends to destroy the structural organization of water and therefore nullifies the thermodynamic driving force tending to form hydrophobic bonds. The latter therefore tend to break down in the presence of organic solvents and the protein molecule is likely to lose its normal conformation and probably its biological activity.
(end of quote from ELMORE, 1968)This quote has served to introduce what factors determine the three-dimensional structure, and thus the functional capacity, of proteins in living organisms. And the content of this quote will be elucidated and further developed (on the basis of subsequent research) further down. In fact we first of all are going to expound the internal structure of liquid water, and then elaborate further on water's accommodation of proteins and on the factors of protein-folding.
The internal structure of liquid waterAs to the "structural organization of water", we must realize that in water hydrogen bonds tend to establish themselves between the water molecules. At subzero temperatures this results in the water molecules arranging themselves in a hexagonal crystal lattice : ice. See next Figure.
Figure above : Structure of ice. Black circles : oxygen atoms. Small white circles : hydrogen atoms. Every oxygen atom is covalently bonded to two hydrogen atoms and hydrogen bonded to a third hydrogen atom. As a result of hydrogen bonds, the water molecules are ordered in a hexagonal lattice and form ice crystals.
Water molecules have an affinity for one another through the hydrogen bond (grey connections between hydrogen and oxygen). A positively charged hydrogen nucleus which 'belongs' to one oxygen atom can still be attracted to the negatively charged electron cloud of a nearby oxygen atom in another water molecule. In ice, this attraction causes the molecules to form a crystalline array similar to the structure of diamond, but not as strong. This very open structure gives ice its low density, and explains why ice floats on water. (After GRIBBIN, "In search of the double helix", 1985)A water molecule, whether in ice or in liquid water, consists of an oxygen atom to which two hydrogen atoms are bonded covalently. The covalent bonds of the two hydrogen atoms to the oxygen atom make an angle of 104.5 degrees. And because the electrons of the hydrogen atoms are sucked to the oxygen atom, the water molecule becomes an electrical dipole : it has a negative electrical charge at the oxygen-end of the molecule and a positive electrical charge at the two hydrogen ends. And this is why water molecules do attract each other electrostatically, they become connected by hydrogen bonds : a positive end (a hydrogen-end) of one molecule attracts the negative end (the oxygen-end) of another water molecule. In this way water forms a hydrogen-bonded network of water molecules. In ice this structure is perfectly present resulting in the hexagonal crystalline form.
In liquid water, which (state) is most relevant and important in the present context of the behavior of proteins in an aqueous medium, there is still something left of this hydrogen-bond based structure :
Figure above : Computer models of liquid water indicate that its hydrogen-bonded network is random and disorderly, and extends throughout the entire collection of molecules like a crazy climbing frame. Here the network is depicted as a lattice of struts representing the hydrogen bonds between molecules. The oxygen atoms of the molecules are located at the junctions of struts. (After HARRINGTON and STANLEY, in BALL, "H2O", 1999)
The hydrogen bond, then, is what sets water apart from other liquids. But it doesn't immediately explain why water is so odd -- why it is denser than ice, why it is central to life, why it has such a capacity to absorb heat, and so forth. Can't we think of water as being just like any other liquid, except more strongly bound?
Not a bit of it. The structure of 'normal' liquids takes rather little heed of the forces of attraction between molecules, [the structure] hinging instead on the packing constraints imposed by the repulsive forces [i.e. in 'normal' liquids the structure (of the bulk liquid) is determined by the molecules adopting a lowest-energy packing pattern, such that their packing is as dense as possible (and then not feeling any attractive forces, if there are any, anymore) but not so dense that they feel the repulsive forces. So in all, in such a packing pattern no net force is felt by the molecules, and this is the most stable packing pattern.]. In water, this little heed of attraction is no longer the case -- it is the attractive forces, the hydrogen bonds, that play the largest role in the way the molecules are arranged. And these attractive forces [much stronger than the usually present van der Waals forces] introduce pronounced preferences for the positions and orientations of neighbouring molecules : each oxygen atom (of a water molecule) sits at the centre of a [irregularly] tetrahedral network of bonds [i.e. each oxygen atom of virtually any water molecule sits, as it were, in the centre of an irregular tetrahedron]. In this respect, water is far less disordered, far more highly structured, than most liquids. It is more akin to a crystal than to a gas. ( BALL, 1999, "H2O", p. 158 ).Today we have good computer models of the structure of liquid water (see Figure above), and BALL, p. 166, tells us the following about them :
So what does simulated water look like? One thing is for sure : it doesn't much resemble ice after all. Forget crystallinity. The network is a jumble, like a collapsed climbing frame. What's more, computer simulations lay to rest the notion, inherent in interstitial models, that there are two classes of [water] molecule, 'ice-like' in the framework and 'free' in the interstices. If that were the case, the majority of molecules would either have four hydrogen bonds on the average, or none. Quite the opposite is found in simulations : most molecules have two or three bonds, and a relatively small proportion has four or none.
Clearly there is a significant rearrangement of hydrogen bonds when ice melts. In particular, whereas hydrogen bonding in ice links each water molecule into rings of six, the most common ring structure in liquid water contains five molecules, not six. And while each vertex of the ice network is tetrahedral, [namely] the confluence of two arms [hydrogen atoms] and two legs [lone pairs of electrons at oxygen], in liquid water some molecules are left dangling -- broken struts on a mangled frame. And a few of the molecules appear to break the rules of the dance by forming five hydrogen bonds instead of four, cheekily grasping two ankles [lone pairs of electrons at oxygen] in one hand [hydrogen atom].
The picture of water structure that emerges from computer simulations, therefore, is neither ice-like nor like a fully random vapour. The molecules form a continuous, disordered and dynamic network of hydrogen bonds in which each molecule is linked with up to four (or very rarely, five) others. Because the hydrogen-bonded network is floppier, more distorted and more defective than that in ice, the molecules are able to occupy some of ice's interstitial space, making the liquid denser than the solid.
[...] This then, is the current [1999] molecular-scale picture of the strange stuff called water : a liquid that has a high degree of internal structure brought about by the 'stickiness' that can bind H2O molecules into a dynamic, ever-changing labyrinth. Water's strangeness resides almost wholly in its hydrogen-bonded character. While it is not by any means the only molecule that can form hydrogen bonds, no other has just the right shape to allow the network to extend throughout all space : the crucial twist of the hips to impersonate water opens up a third dimension. The hydrogen bonds impose structural constraints that are most unusual for a liquid, and these in turn affect the physical properties such as density, heat capacity and heat conductance -- as well as the way that water accommodates dissoved molecules [such as, for us most important, proteins]. And these eccentricities are not just arcane footnotes to the palimpsest of science, but have consequences of global and indeed universal significance.Knowing now something of the internal structure of water, we can now pass on to an exposition of how water accommodates proteins, and how it co-determines the three-dimensional structure and shape of them (And certainly, it is interesting to apply all this to the one protein-like living molecule itself (Unimol), accommodated in the same aqueous medium).
How water accommodates proteinsBefore we continue with Müller's text on inter- and intra-molecular interactions, especially concerning proteins in the living body, we will insert yet another text-part explaining the above exposition of ELMORE, 1968. It is taken from Ph. BALL, 1999 in his book "H2O", A BIOGRAPHY OF WATER, and it is about the role played by water in the biological cell, i.e. how water controls the conformation, the shape, of protein molecules :
Substances that dissolve readily in water are generally polar, meaning that they are not uniformly electrically neutral. If we think of positive charge as blue and negative charge as red, then polar molecules have regions that are blue or red, whereas non-polar molecules are pretty much purple all over. Ions of sodium and chlorine are wholly blue and wholly red balls respectively. Carbon dioxide is blue in the middle, where the carbon atom is, and red at each end, where the oxygen atoms are. Water dissolves polar molecules so well because it is itself polar -- blue at each end [where the hydrogen atoms are], red in the crooked middle [where the oxygen atom is] -- and so confers on the solute molecules a stabilizing attraction between opposite charges.
[note of BALL] To be more accurate, one should say that the affinity of ions [electrically charged atoms or groups of atoms] and polar molecules for water, resides in the latter's high dielectric constant, which is a measure of a substance's ability to attenuate an electric field -- analogous, you might say, to the ability of some materials to damp out sound waves. Because water has a high dielectric constant, dissoved ions of opposite charge do not feel one another's electric field very strongly, and so have less tendency to gather together into a crystal.
Some biological molecules, such as sugars, are also considerably polar and therefore highly soluble in water
Many biological molecules have parts that are polar -- hydrophilic or 'water-loving' -- and other regions that are non-polar -- hydrophobic or 'water-fearing'. Cell membranes are made primarily of tadpole-shaped molecules called phospholipids, which have an electrically charged, hydrophilic head and a non-polar, hydrophobic tail (surfactants such as soap molecules have this same configuration, making them to be able to form colloidal solutions of fatty substances in water, by embedding their hydrophobic tails in insoluble globules of grease.). In cell walls phospholipid molecules are packed side by side in sheets with their heads and tails aligned. The sheets are laminated back to back in double layers, the tails of the molecules in one sheet tickling those in the other (See diagram in previous document and the exposition there about colloids). In this arrangement the hydrophobic tails are shielded from water by the hydrophilic heads. The glue that holds cell walls together, therefore, is not chemical bonding in the usual sense, but the aversion of a part of phospholipid molecules to water. Cell walls are peppered with other kinds of biomolecules, such as membrane proteins that regulate functions like cell-to-cell communication and adhesion, and the transport of ions in and out of the cell.
Proteins have an abundance of hydrophilic and hydrophobic regions -- they are a motley of our metaphorical red, blue, and purple. This is because their amino-acid building blocks have varying degrees of solubility. Surrounded by water, proteins employ much the same strategy as phospholipids : they gather together the hydrophobic parts in a bundle and cover them over with hydrophilic parts. But unlike phospholipids, proteins can conduct this process within a single molecule : the protein chain folds up into a well-defined shape with the hydrophobic regions packed into a buried core and the hydrophilic groups arrayed at the surface, exposed to water.[note of BALL] This tendency to bury hydrophobic parts of the chain should not be overemphasized, however : typically about half of the protein's exposed surface remains hydrophobic. The consequences of exposed hydrophobic surfaces are still controversial.
In this way, the water in which a protein is immersed acts as a kind of cement for the protein's shape : aversion to water helps to guide and secure the folding of the protein chain.
The reason why this is crucial for all of biology is that a protein's behaviour is determined largely by its three-dimensional shape -- by the way it folds. Enzymes, for example, are proteins that act as catalysts for the chemical reactions of the cell : that is to say, they encourage the reacting molecules to come together and interact. A very crude analogy is that of a molecular vice : an enzyme might hold two molecules in the correct orientation to enable them to stick together. The enzyme offers slots, grooves or hollows, called binding pockets, of just the right size and position to hold the molecules in place while the bond forms.[note of BALL] I just make it clear that this is (yet again) an overly simplistic picture by several orders of magnitude. For one thing, enzymes are just as adept at helping bonds to break as to make. For another, they are not really so rigid, and wouldn't function properly if they were. Most enzymes undergo small changes in structure as they bind their target molecules and guide the reaction. And the analogy obscures the important fact that the main role of any catalyst, be it an enzyme or otherwise, is not simply to increase the chances of the reactant molecules coming together with the right spatial disposition, but to lower the energy barrier for the reaction, so that less energy is needed to form the unstable, ephemeral species that appear in the course of the reaction.
The moulding of the peptide chain to create the enzyme's binding pocket is the precondition to its biological role. If the enzyme loses its three-dimensional shape, becoming instead strung out into a looser, floppy chain (a process called "denaturation", since the protein's folded state is called its "native" state), it might as well be just any old polymer, no more able to act as a selective biological catalyst than is a lump of rubber.
Not all proteins are enzymes, but they all need to acquire higly specialized shapes to perform their role in life. The antibodies of the immune system, for example, are proteins that have binding pockets sculpted simply for seizing onto foreign particles in the body, rather than for catalysing their transformation. And the coiled shape of so-called structural proteins in the body's tissues, such as collagen in bone and retinae, or keratine in skin and horn, determines the strength, flexibility and elasticity of the fibrous tissue.
Yet there is no intelligent force to guide the strand of a protein into its folded native state [SHELDRAKE, 1981, 1988, has proposed guidance by morphogenetic fields.]. Rather, the amino-acid sequence in the protein's chain contains the encoded information needed for its proper folding : the protein comes with its own assembly instructions. The blind forces of physics and chemistry are enough to direct the folding process, when the protein is in water. Most proteins don't work at all in other solvents. Implicit in the information programmed into the protein chain is the instruction : "Fold carefully in water". If this is ignored, the information may be misread. And even when the 'instructions' have been followed and the protein has folded to its native state, its usefulness to the cell relies on the continued presence of water. The clustering together of hydrophobic regions of proteins [hydrophobic bonds] is considered by some biochemists to be among the dominant driving force for the adoption and maintenance of a protein's native state.
Yet there are undoubtedly other factors, aside from hydrophobicity, that help a protein to fold properly and to maintain that fold. In particular, hydrogen bonds can form between different parts of the chain, clipping them together. But water molecules can compete for the privilege of sticking to a protein's hydrogen-bonding groups. So there is a delicate balance between the affinity of the protein chain for its own hydrogen-bonding (hydrophilic) regions and for the surrounding water. Loops in a peptide chain can also be secured by strong chemical bonds between the amino acid cysteine, containing sulphur atoms that can hook up together.
But regardless of the relative importance of these and other contributions to the maintenance of a protein's folded shape, it's clear that an understanding of the cell's molecular mechanisms cannot afford to neglect the envelope of water around its molecules. The probelem is, however, that this envelope does some deeply puzzling things. A good way to start an argument amongst cell biologists is to ask them what cell water is like. Let's see what these arguments are about.Everything I've said so far about the question of protein hydration -- the interaction of proteins with water -- has implicitly assumed that we can treat these biomolecules like lumps of putty dumped into a beaker of water, which goes on acting like normal liquid water right up to the protein's surface. Let's think about this assumption for a moment. Water derives its unusual character from its high degree of structure : the molecules are joined into a vast, random network of hydrogen bonds (See Figure above ). Where the network is disrupted by driving a wall through it, the water molecules at the boundary are robbed of their opportunity to form hydrogen bonds with molecules that the wall has displaced. And recall that the network is pervasively cooperative -- one water molecule seizing another by the ankle is sensitive to the wider game of molecular Twister in the vicinity. [The oxygen atom of a given individual water molecule is covalently bonded to two hydrogen atoms, but has still left two "lone pairs" of electrons sticking out. So the oxygen atom with its positively charged two hydrogen atoms (its 'hands') clasps two lone pairs of electrons ('ankles') of two other water molecules, while its own ankles, its two lone pairs of electrons, being clasped by the hydrogen atoms (the 'hands') of two other water molecules. This is the 'twister dance' of the molecules of liquid water.]. With these So the repercussions of the wall's disruptive influence might be felt far afield. In short, there is no justification for treating the water around a lumbering great biomolecule like a proteine as if it were no different from pure water in the middle of a beaker.
So what does water really look like around a protein? It might seem like splitting hairs to be wondering whether this water is or isn't like 'normal' water. But the question goes right to the heart of water's uniqueness as a liquid and how this is manifested in the community of the cell.
One of the conventional ways of describing the hydration environment of a protein [In the Unimol view one might, in addition to the many auxiliary proteins in the Unimol support medium, also think, as to hydration, of the living molecule itself. Also it is embedded in an aqueous medium, the same medium in which all the other molecules of the organismic body are embedded. And the living molecule may provisionally be viewed as a giant protein-like molecule (with many sites of possible hydrogen bonds, and many sites of hydrophilic and hydrophobic subgroups). And indeed, the very shape, the very three-dimensional structure, of this living molecule is co-determined by the interaction of its subunits with the water in its immediate vicinity.] is to assign to the water envelope two kinds of water molecule : 'bound' and 'free'. 'Bound' water is attached quite securely to the protein, typically via hydrogen-bonds [hydrophilic protein-site ==O ... H--O--H (water) ]. These water molecules are not 'bound' irrevocably : any individual water molecule can and does escape rather rapidly. But at any instant the bound water molecules are relatively immobilized -- they are no longer part of the 'true' liquid, their freedom to rotate and tumble is hampered or suppressed entirely. When the protein is crystallized, this bound water crystallizes [also at above-zero temperatures!] with it and typically accounts for nearly half of the crystal's mass. 'Free' water, in contrast, remains largely indistinguishable from water in the liquid far from the surface of the protein : the molecules retain their freedom of movement. Bound water molecules are the bees crawling over a flower's petals, rubbing against the stamen. Free water molecules are those buzzing around the flower head.
This bipartite division of the hydration envelope has some value for cell biologists, but it's far too simplistic. The boundary between 'bound' and 'free' molecules is a blurry one, and even 'free' water near a protein may not be like water 'as we know it' at all.
One of the earliest suggestions that the water of the cytoplasm [the plasm inside the cell, but outside its nucleus] differs from ordinary water came from the Russian scientist A.S. Troschin in 1956. Troschin's idea was given more concrete form in the 1960s by the Chinese biologist G.N. Ling, who suggested that water in the cell forms organized, layered structures on the surfaces of proteins. This bold notion was received sceptically in the West, but succeeded in making explicit the idea that proteins do something peculiar to water structure.
We can break the matter down into two separate questions : how does water accomodate a foreign particle in its midst, and what does water do close to surfaces? Both questions remain contentious.The fact is that nothing -- not a protein, not a sugar molecule, not a mere sodium ion [Na+] from a salt crystal -- can be added to water without shaking up and disturbing its exquisite, dynamic structure. Take the deceptively simple case of a lone positively charged ion like sodium. To get the maximum stabilizing effect from the ion's electric field, the water molecules around it will become reoriented so as to present their 'best' side to the ion -- the side with opposite charge, the oxygen in the cleft of H2O's kink. Water molecules point their rears towards positive ions, but with the best of intentions. This creates a so-called 'primary hydration sphere' of disturbed, reoriented water molecules around the ion, which typically consists of a cordon of between four and eight watter molecules. Water molecules can come and go from this hydration sphere on timescales ranging from small fractions of a second to several hours.
So solute molecules don't just sit immersed in water : they restructure the water in the vicinity. The primary hydratation spheres of some metal ions are virtually crystalline, in the sense that the water molecules are assigned to fixed, geometric positions in space. At first glance, this might lead you to think that all ions enhance the degree of structure of the liquid water in their vicinity. But if an ion disrupts the already highly structured hydrogen-bonded network by forcing some of the water molecules to adopt a new configuration, it's by no means obvious whether the net result is an increase or a decrease in structure. The orderliness in a primary hydration sphere might turn out to be like a jigsaw piece from the wrong puzzle -- it doesn't fit within the jigsaw of pure water.
This consideration has given rise to a distinction between so-called 'structure-making' and 'structure-breaking' ions. Rearrangement of water's hydrogen-bonded network is greatest for small, highly charged ions such as those of lithium, fluorine, and magnesium. The degree of ordering in the hydration sphere around these ions can outweigh the disordering generated in the hydrogen-bonded network further afield, and the ions can be considered structure-makers. On the other hand, big ions with small charge are like the proverbial bulls in a china shop : they mess up the water network but don't offer much local reordering in return. Rubidium, caesium, and iodide ions are structure-breakers. But because it's not obvious how one should add up the structure that is made or broken, water expert Felix Franks says that "much has been written about this concept, much of it misleading".
All the same, structure-making and structure-breaking has some important consequences. It appears to be bound up, in a way that is still unclear, with the effect that salts have on the solubility of proteins. In 1888 Franz Hofmeister noticed that certain ions make proteins more soluble whereas others reduce their solubility. He arranged these ions in an order, now called the Hofmeister series, that reflects their propensity to help proteins dissolve. Highly charged or small ions like sulphate and fluoride -- which are thought to be structure-makers -- promote precipitation or 'salting out', while big, low-charged ions like iodide and perchlorate -- structure-breakers -- promote solubility or 'salting in'. Despite its discovery over a century ago, there is still no satisfactory explanation for the Hofmeister series, although most scientists believe that it is intimately connected with the subtleties of water's hydrogen-bonded structure.
One of the most dramatic illustrations of the effect of water structure on solubility is provided by sugar molecules, which possess hydroxy groups [-OH ] -- an oxygen atom capped with a hydrogen, a fragment of water [itself being H-OH ] welded onto another molecule [ R-OH, where "R" is that other molecule]. Hydroxy groups can engage in hydrogen bonding with the water network. Very subtle differences in the positioning of the hydroxy groups on different sugar molecules can have profound effects on their solubility. The sugar D-talose, for instance, is fairly hydrophobic, whereas D-galactose, which looks barely distinguishable to the casual observer, is far less so. It seems that the flipping of hydroxy groups in D-talose relative to D-galactose makes the former fit less comfortably into the three-dimensional hydrogen-bonded jigsaw of water.Proteins are blotchy amalgams of the red and the blue [the electrically negative and the electrically positive] -- polar, hydrophilic regions -- along with swathes of purple, which correspond to non-polar, hydrophobic parts. To understand how water responds to their presence, we therefore need to know also what water does when faced with purple invaders.
Small, wholly hydrophobic molecules such as methane (CH4), and krypton (a self-contained one-atom molecule) don't form hydrogen bonds, and they don't have electric dipoles that enable them to exploit favourable electrostatic interactions with a polar solvent like water. At face value it would seem that, if you put molecules like this into water, all you are doing is breaking up part of the network of hydrogen bonds to make room for it, without getting any energetic stabilization back in return. No wonder, then, that these gases are not very soluble in water.
But if we look more carefully, we see that this is not really what happens at all. When methane dissolves in water, the water warms up : energy is released. That's a sign that 'bonds' are formed in the broadest sense -- that the methane molecules are welcomed by favourable interactions. In other words, inserting a methane molecule into water's hydrogen-bonded network does not just break bonds without recompense -- there is a considerable payback. Any breaking of hydrogen bonds as the methane forces out a cavity in the network is more than balanced out by other favourable interactions which can only be due to van der Waals forces between the methane and the surrounding water.
And yet despite all this, methane is still pretty insoluble in water. Why so, if it seems to be energetically favourable to surround methane molecules with water? The answer is that the heat change [here the release of heat] is only half the story. The other half concerns entropy, a measure of the disorder in a system. In a crystal all of the atoms are regularly arranged, whereas in a gas they can go anywhere, so long as they don't overlap [i.e. interpenetrate one another]. So there is more disorder -- more entropy -- in a gas, and a substance's entropy increases when it is vaporized.
Whether or not a process of change will occur depends on the balance between the change in heat[note of BALL] Another almost-truth, I fear. Strictly speaking, the balance is between the change in entropy and the change in a quantity called e n t h a l p y, which encompasses heat change due to the making or breaking of chemical bonds but also [encompasses] a contribution that results from changes in volume and pressure -- as, for example, when a piston is pushed out as petroleum is ignited or water evaporates [and volume-increase in a system is work]. The enthalpy change is really a measure of the w o r k that can be extracted from a process, and the motion of a piston (say) is clearly a part of such work.
and the change in entropy that it entails. By "a process of change", I mean anything : the reaction of two chemical compounds, the freezing of water, the toppling of a tree, the formation of a star. The scales that govern all change in the Universe weigh up one of these two quantities against the other.
Methane does not dissolve well in water because the favourable heat change -- the fact that heat is released -- is counteracted by an unfavourable entropy change : the entropy decreases. This tells us that when methane dissolves in water, the two substances are together more orderly overall than they were in isolation. The presence of a hydrophobic molecule like methane amidst the random hydrogen-bonded network of liquid water somehow increases its order.
No one truly knows how to interpret this fact in terms of molecular structure. Indeed, it remains one of the most debated issues in physical chemistry.
For a long time, most researchers believed in a picture of the ordering process that was sketched out in 1945 by Henry Frank and Majorie Evans, which came to be known as the iceberg model. Frank and Evans had this to say :
When a rare gas atom or nonpolar molecule dissolves in water at room temperature it modifies the water structure in the direction of greater crystallinity -- the water, so to speak, builds a microscopic iceberg around it.The rationale for this idea was as follows. Water aims to maintain as many hydrogen bonds as it can. When a non-polar molecule punches a hole in the network, the molecules at the edge of the void have to adopt very specific and rigid orientations to retain all or most of their hydrogen bonds. So water becomes more structured -- perhaps even ice-like -- around the solute molecule. It's like the intrusion of a notorious bore into a crowded party. No one particularly wants to split off from the crowd in his vicinity, or they might be forced to talk with him. So all those around this unwelcome individual carefully turn their backs and engage even more ardently in conversation with their neighbours, standing their ground and adhering to each of their own little group for safety.
Decades after the iceberg model was proposed, researchers came to feel that the term 'iceberg' was potentially misleading. It implies that the proposed solvent cage is both virtually crystalline and composed of ice-like patterns of hydrogen bonds. But if the cage exists at all, it is surely a looser, more dynamic structure. Moreover, rather than being ice-like, formed from rings of six water molecules, the cage might be more like the cavities that form around hydrophobic 'guest' molecules when their solutions are solidified. These crystalline mixtures of water and gas are called gas hydrates, and they bedevil natural-gas pipelines. If the pressurized gas is 'wet', containing water mixed with the methane in the gas field, hydrates can solidify in the pipe and block the flow even above water's freezing point. In gas hydrates the guests are accommodated in polyhedral cages whose faces are made up largely of five-membered rings of water molecules, which are more akin to those believed to exist in liquid water.Theory of hydrophobic bond.
Regardless of the shell's exact structure, one of the implications of the Frank-Evans model is that hydrophobic solutes should attract each other -- that they will cluster together in water. Why is this? Earlier I rationalized the tendency of hydrophobic parts of proteins and phospholipids to stick together in terms of their 'water-fearing' nature -- their relative insolubility. Clustering, I said, is a way of shielding hydrophobic regions from water. But this explanation is oversimplistic, though treacherously seductive. Not only is it circular -- some molecules are hydrophobic because, well, they avoid water -- but it [the explanation, given earlier by Ball] is also nonsensical if we think about energetics alone and neglect the effect of entropy. Methane, after all, gains bonding-type energetic stabilization when immersed in water [energy is released] [so why it is soluble in water so badly?].
In 1959 Walter Kauzmann recognized that the true origin of the hydrophobic attraction and clustering must lie with the entropy of hydration. According to the Frank-Evans picture, which Kauzmann broadly accepted, hydrophobic particles in water are surrounded by hydration shells in which the water molecules are more highly structured, and so have a lower entropy, than their counterparts in the rest of the liquid. If we bring two such particles [or two hydrophobic sites of a single proteine molecule or of different protein molecules] together so that they touch, the hydration shells overlap. This means that some of the constrained water molecules in these shells can be released -- the total amount of water needed to hydrate the two particles is less when they're side by side, just as you can seat fewer people around two tables when the tables are pushed together. In other words, the amount of enhanced water structuring -- of entropy reduction -- imposed by the hydrophobic solutes is decreased when they are brought together, and this clustering will happen spontaneously. It is as if some kind of phantom attraction exists between the particles. This apparent force of attraction is called a hydrophobic interaction, and it arises from the preference for maximal disorder.
Hydrophobic interactions [hydrophobic bonds] clearly do exist -- they keep the bricks of cell walls together and mould proteins -- but it remains uncertain whether they work in the way that Kauzmann proposed, by release of 'structured water'. For all that his idea defined the accepted paradigm for many years, it now appears to have dubious merit -- yet it carries a legacy that is hard to shake off.
Kauzmann's model makes sense only if the hydration shells of hydrophobic particles really do contain more highly structured water. It has proved remarkably difficult to find out if this is so. Small hydrophobic molecules sich as atoms of the inert gases neon, argon and krypton, are the simplest cases to study. But they are so insoluble that it is hard to get enough of them into solution to make their hydration shells detectable in experiments. Measurements made in the past few years do seem to show some ordering of water molecules around inert gas atoms like krypton. But the real question is whether this ordering is any greater than that around a water molecule -- whether the hydrophobic atom enhances the ordering that is already there. The most recent experiments indicate that the hydration shell is not nearly as orderly as the crystalline cages of a gas hydrate, and is probably no more ordered than water in the pure liquid.
But how else can one account for hydrophobic interactions, if not by increased structuring of water? The decrease in entropy -- the loss of disorder -- when hydration occurs might be accounted for partly by the fact that the water molecules in the hydration shell become less free to rotate, rather than because they shift into new, more orderly positions. There is some indication that water molecules around a hydrophobic particle are oriented with their O-H bonds tangential to the particle's surface, as if the water molecules are resting an arm on the particle. Maybe the water molecules thereby accommodate the particle not by increasing their degree of structure but by steadfastly maintaining it : a little reorientation of the molecules could be all that is needed to avoid sacrificing hydrogen bonds [and fewer water molecules will undergo this reorientation when the hydrophobic groups touch one another.]. But here we are in the realm of speculation and can, at present, venture no further.For us, especially in the context of Unimol, it is important to know that the water surrounding the protein molecule (and also the one living molecule) plays -- in addition to the possible hydrogen bonds between (hydrophilic) parts of a protein (or between different protein molecules) -- an important role in moulding the protein, i.e. in determining its three-dimensional structure and shape by way of hydrophobic bonding (hydrophobic interaction). Indeed, hydrophobic interactions clearly do exist and shape protein molecules. Less important for us is to know how precisely water behaves, as to its internal structure, at hydrophobic groups of proteins in order to spontaneously get going the hydrophobic interactions, i.e. to have dF (change of free energy) negative in dH - TdS = dF, where dH is the change in enthalpy (energy difference between initial and final state of the process in terms of work that can be extracted from the process), T the absolute temperature and dS the change in entropy. Even if dH is unfavorable, namely when it is positive (and so contributing to increase, rather than decrease the free energy dF) the entropy of the final state of the process, i.e. the resulting entropy count, may be significantly higher than as it was in the initial state, contributing in making dF negative after all, despite the positive dH. So entropy can make a difference whether a process proceeds spontaneously or not.
But the change in entropy, dS, is a change in the degree of order in a system. And in the present case, where, in order to find out water's precise role in the hydrophobic bond, it is the order of the system of water molecules in the vicinity of a hydrophobic group or surface. And although I -- JB -- am certainly not an expert on hydration, it seems very difficult, if not impossible, to actually and objectively measure the degree of internal order of the water at the vicinity of a hydrophobic site, not necessarily impossible because of the microscopical dimensions or other methodological problems, but simply the impossibility to objectively judge two given different structural states (apart from the purely crystalline state), two structural patterns, of water, whether one has a higher degree of order than the other, or vice versa, in order to calculate the entropy change.
So we stick to the qualitative "explanation" of the hydrophobic bond in proteins submersed in water, by saying that in order to stay away from the water, the hydrophobic regions will cluster together as much they can, and it is this clustering together that is the hydrophobic attraction, interaction or bond. And of course, without the presence of water they will not necessarily do so. So this, then, is the role of water in morphologically moulding protein molecules.In what follows, BALL further discusses the behavior of water near other bodies (atoms, other molecules, proteins). These "other bodies" are presented here as "walls" impenetrable to water. Then these bodies are first discussed when they represent hydrophilic (water-loving) surfaces, and later when they (i.e. other bodies) represent hydrophobic surfaces. And it is the behavior of water, as to its internal structure, that finally may resolve the problem of the precise mechanism of hydrophobic attraction, i.e. the attraction of hydrophobic sites, of a given protein molecule and also of such sites in different protein molecules, to one another. Instead of the Kauzmann model, it is the "drying model" (water evaporating between hydrophobic surfaces close to one another) that accounts for hydrophobic attraction and with it for the moulding of proteins immersed in an aqueous medium. Let's see.
As I [BALL] said earlier, an important issue in understanding how water receives a large biomolecule like a protein, is that the protein presents a physical barrier -- a wall. The molecular-scale structure of any liquid can be altered profoundly when it runs up against a wall. The key consideration here is Desmond Bernal's 'impenetrability' : a wall is a space that the liquid's molecules can't enter. You might imagine that, if there is no force of attraction [to be felt] between the molecules and the wall, the molecules would remain unaware of the wall's presence right up to the point at which they hit it. But that's not so. Close to a wall, a liquid generally becomes organized into layers parallel to the wall.
This ordering of the liquid happens for the same reason that each individual molecule in the liquid is surrounded by concentric shells where the fluid is alternately denser and less dense on average, namely the need for efficient packing of molecules. A wall compels the molecules to pack together more efficiently in its vicinity, creating orderly layers. Given that any large biomolecule has surfaces, we can expect that it will exert a similar structuring influence on the surrounding water. G.N. Ling's early ideas about layering of water at protein surfaces may thus have some validity. Indeed, given that in the cell many of these surfaces are in very close proximity to one another, we must wonder if any of a cell's water is not affected in this way.
Layering of liquids near surfaces happens for any liquid. But you should not expect by now that water will behave just like any old liquid -- and indeed it does not. Water close to surfaces does things that seem utterly baffling, and which remain impossible to explain without stretching credibility to breaking point.
Water near hydrophilic surfaces seems normal enough : it appears to be organized into a series of layers, each one molecule thick. Even this, however, does not go wholly uncontested. Biologist Philippa Wiggins argues that surfaces studded with many charged (ionized) groups [and thus hydrophilic surfaces] -- as the surfaces of proteins and nucleic acids [DNA, RNA] generally are -- sequester a local cloud of ions of opposite charge from the salty cytoplasm, leaving the more remote liquid depleted in ions. This, says Wiggins, sets up an osmotic pressure imbalance between the ion-enriched water near the surface and the ion-depleted water beyond, which in turn makes the former become denser than normal water and the latter less dense and more ice-like. Some studies of water in cells have indicated that indeed it seems able to adopt both higher and lower average densities than ordinary water -- one study in 1984 reported an average density midway between that of normal water and that of ice. Wiggins argues that the 'stretched' water is more ordered and less adept at solvating ions -- in other words, that the concept of water as the ideal solvent for salts might break down inside the cell, where the solvating power of the liquid varies from place to place. It is a provocative idea that would have profound consequences for cell biology if it proves to be true.
But the biggest controversies rage around water at hydrophobic surfaces. In the 1980s, when sensitive methods for measuring the forces between two very close surfaces were developed, evidence began to emerge that two hydrophobic surfaces with water between them attract one another over distances of up to 300 nanometres [1 nm = 10-9 m]. That may sound like a short range -- it is hundreds of times smaller than the width of a human hair -- but it is far, far longer than the range of any known interactions between neutral molecules, which don't extend beyond ten nanometres or so at most. An attractive force extending over 300 nanometres between hydrophobic surfaces in water is almost absurdly long-ranged -- it must be mediated by something like a thousand intervening water molecules. To understand how surprising this is, imagine how you'd feel if you were to be knocked over in Central Station at rush hour by a porter striding past on the opposite side of the hall.
Explanations for this apparent long-ranged transmission of short-ranged forces have tended to focus on water's uniquely structured character. One suggestion is that the hydrogen-bonded network conspires to propagate the layer-like structuring acquired near the surface over many more layers than usual. But there is no evidence for enhanced structuring of water at hydrophobic surfaces beyond a few molecular layers. Another idea is that the long-ranged hydrophobic force is related to the formation of bubbles of dissolved gas beteen the surfaces. When a bubble is wedged between two surfaces, they are linked by a meniscus which pulls them together by surface tension. But the force remains even when the water contains very little dissolved gas.
Alternatively, maybe the water itself just vaporizes between the surfaces. The rationale here is that, while a little rearranging of the hydrogen-bonded network may be feasible around a small hydrophobic molecule like methane [there, there is enough space to accommodate the structural change that took place near the surface of the small molecule], cramming water between two large hydrophobic surfaces is just too destabilizing. The edges of water's jigsaw are disrupted over such a wide area that the rest of the puzzle simply won't fit together in between. Water can't do its dance in too narrow a space. Then the water simply vacates the premises [i.e. leaves the space between the surfaces], leaving only its vapour behind. Because the pressure in the vapour is lower than that in the liquid, the surfaces are pushed together. This phenomenon is called, reasonably enough, 'drying', and calculations indicate that it is expected between hydrophobic surfaces separated by a hundred nanometres or so [So this must then be the hydrophobic attraction, bond, or interaction.].
Whether this mysterious long-ranged attraction between hydrophobic surfaces has any biological consequences isn't known, but that seems entirely possible. Physicists David Chandler, John Weeks, and Ka Lum, suggest that drying, rather than Kauzmann-style hydration, might be the dominant hydrophobic interaction driving protein folding and the aggregation of individual protein molecules into the gangs in which they often operate. The case is far from closed, however, on just how water in narrow spaces acquires its extraordinarily long grasp.
(end of quote from BALL, 1999).
Protein molecules as functional entities
In the above it is attempted (by several researchers) to physically chemically explain the three-dimensional make-up of proteins. In this, as we saw, hydrogen bonds (at hydrophilic sites of the protein molecule) and hydrophobic bonds (interactions) (at the hydrophobic sites of the protein molecule), and thus also the aqueous medium in which the molecule is embedded, do play a pivotal role. And the way these factors work is, as it seems, fully determined by energy exchange and resulting energy states. States or configurations with the lowest potential energy are the most stable and therefore are generated spontaneously (unless some activation energy has to be overcome first). And also on the atomic scale, quantum mechanical energy states determine what happens. In great numbers of atoms and molecules, i.e. in bulk quantities of substances, these quantum mechanical energy states work out in the balance between the thermodynamic quantities of enthalpy and entropy, as explained above.
As such this picture of the formation of the three-dimensional make-up of proteins looks very convincing : A given polypeptide chain with a determined number and arrangement of amino acid residues is embedded in an aqueous medium. And then, as a result of the distribution of hydrophilic and hydrophobic groups in the polypeptide chain, together with the possibility of rotation, in the molecule, about most of the chemical bonds, energetic outcomes will then determine the three-dimensional make-up of that polypeptide chain.
But the resulting protein is not just some chemical substance, but always a truly f u n c t i o n a l entity. The molecule is 'designed' to perform a very specific task, either as materially and morphologically structuring certain parts of the living body, as do proteins such as collagen and keratine, or [the protein performs its task] as an enzyme, catalyzing one or another biologically crucial chemical reaction in the living body, as does an amylase that splits starch or glycogen, or even as enzymes that play a role in the control of protein synthesis itself, or as a hormone, such as insuline which affects the amount of glucose in the blood, etc. Other proteins are adapted to take up, transport, and deliver things, as does hemoglobine, designed to do so with oxygen. And so it is hard to imagine that these highly functional molecules originate as a result of mere thermodynamic or energetical constraints only. These purely physical factors (energy, hydrogen bonds, hydrophobic attraction, etc.) are not functional factors. As to simple molecules, it is true, a given molecule may, in a given organismic context, just turn out to be able to perform some simple function, rendering this molecule functional after the fact. But this cannot take place in the case of very complex molecules such as proteins. They cannot merely turn out to be functional in a certain determined environment, they are functional entities all by themselves. If so, then, in addition to physical factors, there must be a "functional factor" giving the protein its functional three-dimensional structure, a structure, that is, by which it can perform some specific task in the organismic body.
What is this "functional factor"? It cannot reside in the physical material world. This world is not driven by functionality, but by energetic stability only. Well, already as to organismic species, which are in fact functional strategies, we had postulated the "Implicate Oder", an order of reality, that is, which is totally immaterial and in which "interactions" between immaterial forms are "noëtic reactions" or formal derivations of one such form from another. Such forms -- being ontologically, i.e. as to be true BEINGS, incomplete, because they lack matter, i.e. they can only be true beings when they inform matter, or are ontologically carried by matter as a second ontological component of true beings -- strive for becoming material, and this is only possible when they exist and persist in the material order of Reality, the Explicate Order, i.e. when they have been successfully "projected" into that Order. For many immaterial forms this is only possible when they, in the Implicate Order, develop into active "strategies"-to-materially-exist-in-the-Explicate Order, and there they then appear as material individuals of some particular organismic species, i.e. as organisms. Such forms as organismic strategies are, of course, very complex forms. But there also are forms in the Implicate Order that can directly be projected into the Explicate Order, without themselves being an active strategy and appear as inorganic substances wherever and whenever they turn out to be energetically stable. And, further, there are, qua "size", intermediate forms that cannot as such materially exist in the Explicate Order but can only exist there as part of an organismic strategy. And these are, among others, the forms of biomolecules such as nucleic acids and especially proteins. So as a result of being an enzyme or other functional protein, the forms representing them can be projected, and then materialized, into the Explicate Order, and appear there in the appropriate organismic bodies. So protein molecules are in fact materialized strategies within higher-order materialized strategies. And such molecules may even be artificially synthesized and materially exist in some simulated environment, that is, in certain laboratory conditions. Indeed, their existential condition is much less demanding than that of a full organismic strategy, an organism.
So proteins in their native condition are folded molecules and as such being able to perform some biologically important function. And because the folding is functional, i.e. aimed at a functional structure, it cannot be the consequence of solely its amino acid sequence, because that in itself is not something functional. But, above we had discussed theories physically trying to account for the folding of proteins, involving energy, hydrogen bonds and hydrophobic attraction. But, as we have seen, these theories are not fully capable to rigorously explain the folding phenomena. Indeed, protein folding is a major problem in physical chemistry. And the reason for this may be that all energetic and physical factors are simply not enough to explain the folding, i.e. they, all by themselves, cannot accomplish the folding. They can, of course, result in some simple folding of teh peptide chain, but not in a very specific and complex functional folding. In this, non-physical factors must play a role, as we expounded above. In order to support this conclusion it is perhaps instructive to quote [and paraphrase] a section or two from R. SHELDRAKE'S book "A New Science of Life", 1981, where he discusses the problem of reductionistically (here quantum mechanically) deducing the structure of molecules, and here especially that of the complex folded molecules of proteins as they are found in living bodies.
The prediction of chemical structures
Quantum mechanics is able to describe in detail the electronic orbitals and the energy states of the simplest of all chemical systems, the hydrogen atom. With more complicated atoms and with even the simplest chemical molecules its methods are no longer so precise. The complexity of the calculations becomes formidable, and only approximate methods can be used. For complex molecules and crystals detailed calculations are impossible, at least in practice. The structure of the molecules and the atomic arrangements within crystals can be found out empirically, by chemical and crystallographical methods. These structures may indeed be more or less predictable by chemists and crystallographers on the basis of empirical[ly established] laws. But this is a very different matter from providing a fundamental explanation of chemical structures by means of the Schrödinger wave equation.
It is important to realize this severe limitation of quantum mechanics. Certainly it helps to provide a qualitative or semi-quantitative understanding of chemical bonds and of certain aspects of crystals, such as the difference between insulators and electrical conductors. But it has not enabled the forms and properties of even simple molecules and crystals to be predicted from first principles. The situation is even worse with regard to the liquid state, of which there is still no satisfactory quantitative account. And it is illusory to imagine that quantum mechanics in any detailed or rigorous way explains the forms and properties of the very complex molecules and macro-molecular aggregates studied by biochemists and molecular biologists, not to mention the vastly greater complexity of form and properties of even the simplest living cell.
So widespread is the assumption that chemistry provides a firm foundation for the mechanistic understanding of life, it is perhaps necessary to emphasize on what slender foundations of physical theory chemistry itself rests. In the words of Linus Pauling [the "Einstein of chemistry", who quantum mechanically has sorted out the nature of chemical bonds], 1960 :
"We may believe the theoretical physicist who tells us that all the properties of substances should be calculable by known methods -- the solution of the Schrödinger equation. In fact, however, we have seen that during the 30 years since the Schrödinger equation was discovered only a few accurate non-empirical quantum-mechanical calculations [i.e. predictions] of the properties of substances in which the chemist is interested have been made. The chemist must still rely upon experiment for most of his information about the properties of substances."Although a further 20 years have passed since this passage was published, and although there have been important improvements in the approximate methods of calculation available to quantum chemists, the situation remains essentially the same today [1981].
Nevertheless, it may be argued that the detailed calculations could be carried out in principle. But even assuming for the purpose of arguments that these calculations could indeed be performed, it cannot be known in advance that they will be correct, that is to say agree with empirical observations. So at presence there is no evidence for the conventional assumption that complex chemical and biological structures can be fully explained in terms of existing physical theory.
The reasons for the difficulty, if not impossibility, of predicting the form of a complex chemical structure on the basis of the properties of its constituent atoms [and in the case of proteins, on the basis of their amino acid sequence] can perhaps be understood more clearly by means of a simple illustration :
Consider elementary building blocks which can be added to each other one at a time either endways or sideways. See next Figure.
Figure above : Possible combination of different numbers of building blocks capable of being joined together either endways or sideways. (After SHELDRAKE, 1981)
With two building blocks there are 22 = 4 possible combinations. With three, 23 = 8. With four, 24 = 16. With five, 25 = 32. With ten, 210 = 1024. With twenty, 220 = 1048576. With thirty, 230 = 1073741824. And so on. The number of possibilities soon becomes emormous.
In a chemical system, the different possible arrangements of atoms have different potential energies owing to the electrical and other interactions between them. The system will spontaneously tend to take up the structure with the minimum potential energy. In a simple system with only a few possible structures, one [of such structures] may have a distinctly lower energy than the others.
Figure above : A diagrammatic representation of the possible structures [varying along the horizontal axis of the diagram] of systems [such as molecules] of increasing complexity [A, B, C, D]. [the energy content of a structure varies along the vertical axis of each diagram]. In A, and also in B, and C, there is a unique minimum-energy structure [i.e. lowest energy is possessed by only one structure], but in D several different possible structures are equally stable.
(After SHELDRAKE, 1981)In A (of the above Figure) this [simple system] is represented by the minimum at the bottom of the 'potential well'. Other less stable possibilities are represented by local minima on the side of the 'well'. In systems of increasing complexity, the number of possible structures [of these systems, of these molecules] increases (B, C, D, in the above Figure). As it does so, the chance of there being a unique minimum-energy structure seems likely to deminish. In the situation represented by D, several different structures would be equally stable from an energetic point of view [in the same circumstances]. If the system were found to take up any of these possible structures at random, or if it oscillated between them, then there would be no problem. But if it invarably took up only one of these structures, this would indicate that some factor other than energy somehow determined that this particular structure was realized rather than the other possibilities. No such factor is at present recognized by physics.
Although chemists, crystallographers and molecular biologists cannot carry out the detailed calculations necessary to predict the minimum-energy structure or structures of a system a priori, they are able to use various approximate methods in combination with empirical data on the structures of similar substances. In general, these calculations do not permit unique structures to be predicted (except for the simplest of systems), but only a range of possible structures with more or less equal minimum energies. Thus these approximate results appear to support the idea that energetic considerations are insufficient to account for the unique structure of a complex chemical system. But this conclusion can always be avoided by re-asserting that the unique stable structure must have a lower energy than any other possible structure. This assertion could never be falsified because in practice only approximate methods of calculation can be used. The unique structure actually realized could therefore always be attributed to subtle energetic effects [causing it to be the lowest-energy structure] which eluded calculation. The following discussion of Pauling's (1960) illustrates the situation with regard to the structure of inorganic crystals :
"Simple ionic substances such as the alkali halogenides [for example NaCl (table salt), KCl, CsCl, KBr, NaF, etc.] have little choice of structure. And a very few relatively stable ionic arrangements corresponding to the formula M+X - exist, and the various factors that influence the stability of the crystal are pitted against one another, with no one factor necessarily finding clear expression in the decision between the sodium chloride (NaCl) and caesium chloride (CsCl) arrangements [i.e. crystals having arranged their (positive and negative) ions as they are in NaCl or as they are in CsCl]. For a complex substance, such as mica, KAl3Si3O10(OH)2, or zunyite, Al13Si5O20(OH)18Cl, on the other hand, many conceivable structures differing only slightly in nature and stability can be suggested, and it might be expected that the most stable of these possible structures, the one actually assumed by the substance, will reflect in its various features the different factors which are of significance in determining the structure of ionic crystals. It has been found possible to formulate a set of rules about the stability of complex ionic crystals [...] These rules were obtained in part by induction from the structures known in 1928, and in part by deduction from the equations of crystal energy. They are not rigorous in their derivation nor universal in their application, but they have been found useful as a criterion for the probable correctness of reported structures for complex crystals and an aid to X-ray investigation of crystals by making possible the suggestion of reasonable structures for experimental test."The range of possible structures becomes much greater in organic chemistry, especially in the case of macro-molecules such as proteins, the polypeptide chains of which twist, turn and fold into complicated three-dimensional forms. See next Figures.
Figure above : Upper image : The structure of the enzyme phosphoglycerate kinase, isolated from horse muscle. alpha-helices are represented by cylinders and beta-strands by arrows.
Lower image : The structure of an alpha-helical region in more detail.
(After BANKS et al., 1979, in SHELDRAKE, 1981)Figure above : Diagrammatic representation of the three-dimensional structure of four kinds of protein molecules.
(From Advances in Protein Chemistry 34 by J.S. RICHARDSON, 1981, in SHELDRAKE, 1988)Figure above : Hierarchical levels of protein structure.
The bimolecular complex shown here is of the catabolite activator protein, which plays a role in the control of protein synthesis in the bacterium Escherischia coli by binding to DNA.
(From Advances in Protein Chemistry 34 by J.S. RICHARDSON, 1981, in SHELDRAKE, 1988)There is good evidence that under conditions in which a given type of protein molecule is stable, it folds up into a unique structure. In numerous experimental studies, proteins have been made to unfold to varying degrees by changing their chemical environment. They have then been found to fold up again into their normal structure when they are replaced in appropriate conditions. In spite of starting from different initial states and following different 'pathways' of folding, they reach the same structural end-point (ANFINSEN and SCHERAGA, 1975).
This stable end-point is likely to be a minimum-energy structure. But this does not prove that it is the only possible structure with a minimum energy. There may be many other possible structures with the same minimum energy. Indeed, calculations designed to predict the three-dimensional structure of proteins, using various methods of approximation, invariably give far too many solutions. In the literature on protein folding, this is known as the "multiple-minimum problem".
There are persuasive reasons for thinking that the protein itself does not 'test' all these minima until it finds the right one :
"If the chain explored all possible configurations at random by rotations about the various single bonds of the structure, it would take too long to reach the native configuration. For example, if the individual residues of an unfolded polypeptide chain can exist in only two states, which is a gross underestimate, then the number of possible randomly generated conformations is 1045 for a chain of 150 amino acid residues (although, of course, most of these would probably be sterically impossible ones). If each conformation could be explored with a frequency of a molecular rotation of 1012 [rotations] per second, which is an overestimate [i.e. the frequency of the molecule trying out each intra-molecular rotation, is in fact lower, making the whole test even last longer], it would take approximately 1026 years [for the molecule] to examine all possible conformations. Since the synthesis and folding of a protein chain such as that of ribonuclease or lysozyme can be accomplished in about two minutes, it is clear that all conformations are not traversed in the folding process. Instead, it appears to us that, in response to local interactions, the peptide chain is directed along a variety of possible low-energy pathways (relatively small in number), possibly passing through unique intermediate states, towards the conformation of lowest free energy." (C.B. Anfinsen and H.A. Scheraga, 1975)
[Where we [JB] would like to add or rather anticipate : [towards the conformation of lowest free energy] namely [the chain being] directed to that particular conformation of lowest energy instead of to some other conformation of (about) the same lowest energy. That is to say, in folding, the polypeptide chain is not only directed straight to some lowest energy conformation, but to a particular one of them. And upon renewed unfolding, it will, in appropriate conditions, fold back again to precisely this conformation. And further we must stress that this particular conformation, the native state of the protein, i.e. its natural folded state, may not only be not unique, but even not a lowest-energy conformation at all. It could be merely metastable (meaning that having it to fall towards the very bottom of the potential energy well, its needs a push, i.e. it needs to be perturbed, or it can't reach this lowest-energy strucure at all) : [Perhaps] "the observed folded state is not the most stable thermodynamically of all those possible, but merely the most stable state of those which are kinetically accessible" (CREIGHTON, 1978).][Sheldrake commenting on the above quotation] But not only may the folding process be "directed" along certain pathways, it may also be directed towards one particular conformation of minimum energy, rather than any other possible conformations with the same minimum energy.
This discussion leads to the general conclusion that the existing theories of physics may well be incapable of explaining the unique structures of complex molecules and crystals. They permit a range of possible minimum-energy structures to be suggested, but there is no evidence that they can account for the fact that one rather than another of these possible structures is realized. It is therefore conceivable that some factor other than energy 'selects' between these possibilities and thus determines the specific structure taken up by the system.
( end of quote from SHELDRAKE, 1981 )
Sheldrake explains this "other factor" by having it to be a non-physical field guiding the folding process and stabilized by repeated occurrence of folding along a particular pathway resulting in a particular conformation. We [JB] on the other hand, focus rather on the functional aspect of natural proteins. The "other factor" then is the very function the protein has to fulfill, and is at work in the Implicate Order, resulting -- after projection into the Explicate Order -- in a functioning molecule inside an organismic body forming its existential condition.
Of course the earlier mentioned possibility of forming hydrogen bonds and realizing hydrophobic attractions between parts of the same individual protein molecule, are important factors realizing protein folding. And because they in turn depend on the specific amino acid sequence in such a protein molecule, we may reduce all physical and chemical factors of protein folding to the protein's amino acid sequence. But, we have seen that this is not enough for having proteins folding up into unique but specific three-dimensional, and moreover functional, forms. For a fine example, let us quote a report from SHELDRAKE's 1988 book, The Presence of the Past :
The haemoglobins provide an even more extreme example. These red proteins are responsible for the colour of the blood, and are found in a very wide range of animals, both vertebrate and invertebrate. Even peas and beans produce haemoglobin : it is present in their root nodules, which is why the nodules are pink inside. The three-dimensional structures of these various kinds of haemoglobin are extremely similar. However, their amino acid sequences are quite different. In all known haemoglobin sequences, only 3 out of a total of 140 to 150 amino acids are the same in the same positions.
Such an extraordinary stability of structure in spite of differences in amino acid sequence is astonishing if we assume that all the information required for the folding of the protein chain is contained in the amino acid sequence.
(Continuing with the text of Müller, 1959, (Unimol). See HERE where we interrupted this text)
The idea of molecular compounds (MC) is perhaps also evident by the fact that the bonding conditions (just as in proteins) cannot simply placed into some classification -- a fact already pointed to by Pfeifer -- "the free residual affinity, as a lower degree of unsaturatedness, having to be complemented by a special tuned-to-one-another of the components". The (probably best to be described quantum theoretically) phenomena of stabilization in MC on the basis of mutual orientations and special constituent configurations, give an -- qualitatively certainly very pale -- image of the vital-stabilization.
The question, whether the principle of MC does play a role in the organismic domain may perhaps be answered as follows : as addition and completion it surely plays a role, whereas essentially and chief-functionally it does not, and this [latter] opinion being derived from analogy, because generally in a semi-liquid medium there is only a lowered degree of stability, while in fact great stability is required. The loose bonding [in MC] can perform too little. It is much better functionally replaced by the true full-valence bond, providing the most reliable solution of all assumptions of special stabilizing forces in all systems and in all possible conditions. So, the "forces", active in MC, probably are in one or another form also active in the vital domain, but certainly only casually and completing so, or "built in", but not as replacing [the other forces].
Müller now inserts an exposition on so-called "encasing molecules" and their comparison with symbiosis and parasitism. After it, he returns to molecular compounds (MC) again.
On encasing molecules
Because in the past decennium [1949-1959] a particular form of molecular compounds (MC), namely the so-called encasing compounds (EC), were much discussed, we would like to mention them here, especially because they let their concept to be extended until including certain biological facts.
With an EC (encasing compound) we mean the fact that a mostly larger so-called host-molecule possesses (or takes up) a spatial structure in which a ring-like, spherical, or channel-like hollow is present in which [hollow] relative weak but additive intermolecular forces are active, and which [hollow] can, stoichiometrically and stably, receive a suitable guest-molecule while holding it there by encasing it. Surmising that super-polymers from the haemin- and chlorophyll-series may contain encasing sites, and that proteins may form EC (ferment models) ( NOTE 859) gained interest, when it was found that alongside the first known and only solid EC's splitting up in solution, there were also EC (cyclodextrine) which also in solution preserve the hollow, and holding guest-molecules ( NOTE 860).
It is understandable that we, as a result of the eminent problem-solving ability of Unimol, do not welcome the EC with such a hopeful enthousiasm as do many authors working with this interesting phenomenon. Nevertheless we take them to be truly remarkable.
Because one may grant proteins -- insofar we have not to do with fixed immobile structures -- already the ability to form EC, the in itself not yet life-specific system of EC is possibly an auxiliary feature, either as to the formational pathway or as permanent support of the true construction-by-bonding. In exploitating such and other phenomena, the bond and the resulting configuration may still enjoy a little freedom. Following the concepts "inner-complex formation" and "inner-salt formation" one may possibly speak of inner EC within its own molecule. It may be a sort of (mechanical) stabilization in itself as one [case] among many others.
To us the EC appear to be remarkable therefore because they seem to show a principle of "intimate" system formation or also the archetype of a "mechanical" symbiosis (with the "aim", beneficial to both system partners, of an energetic and thus of state-stabilization), that we particularly appreciate.
Symbiosis provides an example of the fact that an illustrative concept, derived from zoology and botany, may formally -- and with, on the one hand, a little caution and an unavoidable extension or also reduction on the other -- be applied to basic phenomena, from which one never would have derived this concept.
Starting from "above", we first have the close coexistence of separate metazoic or metaphytic organisms. Then follow intracellular symbioses ( NOTE 861), in which the partners in themselves are still separate and separable, i.e. also being free-living, organisms. Then follows a more or less doubtful domain of intracellular symbioses of cells and plastids [Small, variously shaped bodies in cytoplasm of plant cells (excluding bacteria, blue-green algae, fungi, slime-fungi), one to many per cell in different plants, containing pigments and/or reserve food materials.], in which both partners cannot freely exist anymore. Subsequent to them [i.e. still further going down the ladder of size] one arrives at an up to now speculative domain of the most extensive molecular symbiosis (with ferments) with pronounced historical components.
Finally, one leaves the organismic domain and is in the inorganic. The aggregation, taking place there, from molecules to molecular compounds and crystals, crystallites, micelles, etc., and the aggregation from atoms to molecules, also may entirely be placed under the concept of symbiosis ( If one, generally, has given this up, then less so by reason of contradictions, but especially because such way of seeing things doesn't offer any profits).
The formal symbiotic benefit (this , of course, has to be verifiable in one way or another) may boil down to assume energetically more stable existential forms. This also holds for the molecularly bound form of atoms, which now, when tied up in some molecule, are "protected" from being attacked by other partners and becoming transformed, i.e. losing their special present existence. But this also holds for the molecules which, in the most simple case, namely crystallization, assume the most stable form. And it especially illustratively holds for molecular compounds (MC), whose existence is based on the aggregation, and among them in turn, most strikingly, for encasing compounds (EC), in which many of the host partners assume a definite form and with it a definite configuration not until having come under the influence of guest partners (here it is about entirely regular molecular symbioses). Takes one this special form as to be the [compound's] individual existence (finally even as "aim" or "purpose"), then it owes this special form exclusively to the presence of the partner, and stands or falls with its presence. So the symbiotic image fits well, and it is more than some abstruse form of description of interactions, a description having originated from some obscure natural philosophy ( NOTE 863). The partly quite positive heat gradation in EC [i.e. the release of energy during the formation of the EC] provides thereby the appropriate energetic counterpart.
If one doesn't allow to be embarrassed by this peculiar way of seeing things [i.e. speaking about molecular "symbiosis"], then one sees that the most expressed and most effective symbiotic forms are forms where chemical bonds are involved (also among EC there are such symbiotic forms in which undoubtedly chemical forces act between the partners). This criterion of appropriateness certainly is independent of the [organizational] stage, so that also in higher systems (living substance) there is the most effective "symbiosis" only when chemical bonding is involved.
( Continuing with Molecular Compounds (MC) )
First orientational component in protein molecules.
Against the formation of molecular ions act the Coulomb forces of repulsion between equal electrical charges [i.e. charges of the same sign], which [forces] certainly become small in aqueous solutions because the high dielectrical constant of water renders these repulsive forces to become small, while the attractive [coulomb] forces [attraction between particles having a charge of different sign] are independent of the static dielectric constants. The positively attracting forces are, of course, partly also, effects of molecular ions (having to do with the complementary charge) on others, [the positively attracting forces] being the inter-molecular forces of cohesion resulting from the polar structure which is, as to charge, compensated towards the outside.
One here may give up on the somewhat model-like viewed "polar" structure, and suppose that proteins offer further non-chemical possibilities for association, such as for instance quantum mechanical resonance-stabilizing forces of attraction, which attraction, insofar as it is about entirely equal molecules, is only superior to van der Waals or field-valence forces ( NOTE 863a - note of author of website) of unsymmetric multipolar compounds, but at the same time only [so superior to van der Waals forces] into the direction of in any case to be avoided crystalline aggregation ( NOTE 864). One may avoid the possibility of crystalline aggregation most reliably [by doing it] chemically. Super-molecularity [i.e. large molecules] alone is certainly not sufficient, because the very commoness of crystalline fibers is a general macromolecular-physical phenomenon. It [i.e. avoiding crystallinity] therefore needs an additional factor :
The structure of proteins -- whereby we here do not think of the native "crystallizable" product proteins, but of the protoplasmatic content-substances resisting any isolation into their pure state -- with its many amino acid components, as well as with its various (for instance glucosidic) side chains and its possibilities of forming isomers, shows a great sequential irregularity and correspondingly a low degree of [translational] periodicity, so that -- what, as has been said, must be avoided in any case -- the inclination to form crystalline associations is vanishingly small. So given that the danger of a crystallogeneous condition [a condition allowing crystallization] in the protoplasm, a condition which definitely does not represent the direction towards the living condition, is minimal, one, on the other hand, does not have a foothold that the protein inter-molecular forces of cohesion alone are able to generate the zoogeneous [i.e. living] condition ( NOTE 865) (in which we always depart from the general conviction that the individual molecules for themselves do not yet live, - because when they do not yet live the interactions can [without disadvantage] be arbitrarily loose, and life only appears in the [chemical] combination .
So one can say : The -- insofar as a corresponding strong spatial approach is given ( NOTE 866) -- always and automatically between neutral molecules occurring and superpositioning interactions through polar-, induction-, and dispersion-forces, may, it is true, result in diverse loose, and under special circumstances also temporarily stable, molecular associations. But even among special protein molecules the normal inter-molecular forces in all their diversity in size and kind nevertheless show a certain uniformity. And be it that the known relationships of interaction are physically and mathematically not entirely clear, and that we may expect additional unknown factors, there nevertheless is no empirically verifiable indication that as a result of the mentioned relationships between free molecules [i.e. between free molecules that remain free] qualitatively could originate such a thing that we call life. In [mere] colloid-chemical conditions of living substance we do not see any possibility such that here would appear such stable relationships allowing a constant impeccable function.
All what is observed until now, also the most surprising gel-systems, are in this respect "empty" and show -- even when they should play a practical role in the organismic condition -- no foothold for specific, namely life-specific self-directing, coordinated and cooperatively reacting and finally "self-recognizing", possibilities. If one is not prepared to supplement the, in higher organisms increasingly necessary, principle of contact [accomplished] by the free chemical messenger substances bridging the overall molecular distances, with a nonsensical legion of additional messenger substances realizing the functional contact between the uncountable small system-molecules (which legion spatially and qua number would be of the same order of magnitude as is that of the mentioned small molecules ! ), then one must replace the inter-molecular small power of performance, which not even can resist the normal thermal motions ( NOTE 867), by the much more effective inter-atomic forces. This reduced framework-consideration is valid for all combinations and systems of molecules, so that we therefore may say that there cannot be any system of free "protein" molecules constructed through inter-molecular or similar forces, a system such that it constitutes life.
Second orientational component in protein molecules.
Let's go, therefore, to the second orientational component of protein molecules. This consists in the various groups [in protein molecules] which can chemically react with one another, and which are thus capable to connect two protein molecules with each other at one or more sites [of attachment], and connect so strongly ( NOTE 868) that this new form -- a double-molecule, that is, a double-sized one-molecule -- is invariably stable, because the firmness of the chemical bond, a firmness in special cases moreover be supported by the formation of micelles, can stand up against effects that interfere with permanent aggregation, for example can stand up against the sometimes violent thrusting of the molecules of the solvent and against the Brownian motion of dispersed particles, and thus the double-molecule will persist. Because this consideration doesn't need to stop after the double-molecule (See the Section on the renounced dimensions), but may well be continued up to the fine-histological [i.e. the domain of cells and tissues], and finally even up to anatomical structures, because to this there are no fundamental objections, and because in this way all demanded tasks and functions are uncomparably well and confidently solved, we say that the inter-molecular relationships (in the true living substance), necessary in the system of the living, must go through true bonds, and that thus the alleged system of free protein molecules is "in reality" a single truly chemically bonded macro-molecule.
This certainly isn't a premature conclusion, and we, in our consideration, do not want to ignore what an enormous type- and reaction-expressing role is played by the (not truly bondingly) "physical" inter-molecular forces in our world, yes, that its actual existence for the most part rests upon them. Only the special state of the living doesn't fit this simple formula ( NOTE 870). It will also be wise not to sharply distinguish between the old classical valences and residual bonding forces. The differences should not be artificially erased, but they erase themselves naturally, and effective is always their sum or combination. In some cases, the sideline forces, finally still experiencing a strengthening compensation, may be in a stronger degree quality-determining than are the so-called chief bonding forces.
Here, we once and for all, lay down, that there are, so to say, many ways all terminating at the same end-point, so that a convergence becomes visible, giving us the assurance of having taken the right path, and of possessing a truly usable and strong foundation for our all-encompassing system of clarification.
Belonging, among other things, to the above mentioned convergence is the fact that we must formulate the impression or assumption of special inter-molecular forces in such a way that they, by the above mentioned reasons, have the rank and magnitude of the intra-molecular "forces", that they, therefore, are practically not distinct from them, i.e. that they are of the same nature, and that the molecular boundaries lie precisely there where these inter-forces leave off, and this again means a single (higher-level) macro-molecule ( NOTE 871).
In specifying our special view we do not, however, want to say that basically pre-formed micro-molecules are, or become, larger molecules by way of chemical apposition, but that, the other way around, these larger molecules are formed right from the beginning and exclusively so, but that they are physiologically or experimentally capable of disintegrating into smaller fragments, which [fragments] -- after simultaneous moderate transformation -- are independent, and are identical with, or similar to, known organic molecules.
The experimentally found unequivocal continuous transition of nerve ends into the (muscle) cell, supports the cell-potential-wholeness view, and in this sense it is also a necessary demand if one considers the development of life from its very orgin. How would the stimulus-reactive perceptively-acting living molecule in its primordial state disintegrate into molecule fractions, or form doublings, that is, molecule fractions [still] working holistically together in specialized forms? One may think as much as one wants of animal associations [such as herds and colonies] or of insect societies with division of labor, but if one wants to work with this image then also here one automatically arrives at the demand for a relationship which completely matches up with the chemical bonding relationship. [i.e. if we think of how to interpret the cooperation between the constituents of an individual organism, we are inclined to think of this cooperation as it is in herds, colonies, and insect societies. But the cooperation within a single organism cannot be equated with that in the mentioned animal associations, it is much closer, and the cooperating constituents (in an individual organism) have much less individuality than the members of an animal society have. And according to HOENEN's metaphysical theory of the molecule, of any molecule, the constituents of it are not material particles at all (let alone individuals) but virtual particles, meaning that they are qualities of the molecule. They are not independent but intricately connected through the molecule as a whole, there is a close relationship between the qualities of the molecule. And this indeed suggests that also an individual organism is a single molecule, and the bonding relationship within an individual organism, i.e. between its "constituents", must have the strength and nature (and specifity) of the chemical bonding.]
One should well understand that we, with our organismic one-molecule, a molecule for many orders of magnitude surpassing the micro-molecule, do not refer to some big coarse lump of plasm (as we might, analogously, think of a macroscopic crystal being indeed a single molecule but not the one-molecule we have in mind, or we might think about a macroscopic polymer consisting of simple and repeated units, which we also do not have in mind), but a, it is true, very extensive form, as to its boundaries covering those of the organismic-morphological, and fixed by the continuous bonding relationship, a form provided with all the important characteristics of the one-molecule.
In order to further characterize things, we may say : The living substance is a strongly hydrated one-colloid ( NOTE 872) of unknown fine-structure, being protein-equivalent, with uninterrupted bonding, with apposited (deposited) or inserted labor-substances, mechanical encasings (backup substances, and others), and especially [the living substance] being the carrier (the substrate) of all life-characteristic features ( NOTE 873). Living matter neither possesses a sufficient inner sequence-periodicity ( NOTE 874), nor a -- realizable through parallel shifts (translations) -- building block periodicity, to succumb to the contra-vital crystalline order ( NOTE 875, important note, supplemented by a further theoretical discussion). This danger only exists in the case of "pure" metamerization or the simple parallel repetition of precisely the same structural order. An image of the latter probably might be the non-living cryastalloid TMV-shell-protein [TMV = tobacco mosaic virus], consisting of many "parallel" placed polypeptide chains with the same end-groups and probably having the same amino acid sequence. Such substances can always "crystallize".
The special structural principles of living substance are -- considered as to the purely chemical substance -- not opposing one another, but, as to constituents, specifically possible ones and making possible the life-characteristics. To emphasize a dynamical structure of the living ( NOTE 876) as compared to the static structure of the inorganic-crystalline order (including the true amorphic), means the over-evaluation of a [mere] auxiliary function. There, where true dynamics is involved, namely within the atomic domain [i.e. within a molecule], it is precisely the same in the inorganic as in the organismic domain.
There are neither living systems no living orderings or organizations, but only living true compounds, living molecular substances [alongside non-living molecular substances], which, it is true, normally need the environment in which they once have originated and now continue to live in, and therefore appearing together with it, a phenomenon being responsible for the view of a living system. These molecular compounds are at the same time living organisms. The Unimol bonding-combinations are in their entire range physical individual forms, which must functionally and existentially rely on a medial system, but not identifying with it to such an extent that one must emphasize the system. To be emphasized is only the uni-molecular system [the uni-molecular whole].
In order to picture the overall state of affairs, one may reduce the organismic mega-molecule to a single polypeptide chain [a chain of amino acid residues intra-connected by peptide bonds, and forming the backbone of proteins]. Thinking in terms of a model, then, in virtue of purely group-chemical considerations, we may assume in such an entangled polypeptide substantial inter-molecular forces. On the one hand, as a result of too small a periodicity -- and so all in all comparable with a mixture of purely distinct, but nevertheless similar molecules -- no crystalline aggregation can take place, and as a result of the strong effectiveness and of the special states of resonance, on the other hand, a permanent close-range orientation can appear, whose interaction is so strong that it can resist the disorienting forces. Because it is about one single molecule, the inter-molecular forces are at the same time intra-molecular forces of aggregation ( NOTE 877), which simultaneously add up with the purely chemical bond, and sometimes being able to "mix up" with it ( NOTE 878). A simular consideration does apply to several polypeptid chains [instead of one], which can truly bond to each other and intra-molecularly strongly aggregate ( NOTE 879).
The organismic mega-molecule does represent the special case in which the not entirely uniform, inhomogeneous medium of dispersion [the aqueous support medium] is partly bound [to the mega-molecule] in such a fashion that may at most be seen as a special case of inter-molecular forces approaching true bonding ( NOTE 880). Perhaps this system is even stepwise organized, namely : A kernel of uninterrupted true bonds of living substance, then a "layer" of apposed [adhered-to] macro-molecules of various kinds (at least partly protein-like), then a layer of hydratically bound water -- perhaps covered by a swarm-like oriented water layer -- and finally the open medium, the aqueous, containing or forming ions, medium of dispersion. This structure must, of course, be imagined microphysically, and [thus] not seeing in it, say, the structural principles of the cell ( NOTE 881). The very important plasmatic boundary surfaces oriented towards the open media are formed by specific and probably semi-permeable denaturation membranes, which, as a result of inclusion of phosphatides, fats, etc. are sealed up, resulting in mechanical and chemical barriers. Because this ability is a very old craft, the formation of "naked" (but always having membranes) cells was not such surprising an accomplishment where all the rest was already a one-molecular whole. [Because in such a whole its boundaries are present anyhow, without being separate structures.]
The organismic one-molecule can, at all conceivable places, achieve microphysical fineness (delicacy), and conclusions from it may be drawn at all times to begin with. But insofar in this fineness possibilities of disturbance do exist, one must also assume that functionally and selectionally such possible disturbances may be compensated for or neutralized, which is perhaps the main part of the organismic burden [the organism is a dissipative structure anyhow, constantly getting rid of (order-destroying) entropy and waste material.]. Metamerizations [formation of metamers, sequential pieces, especially in organisms] of many kinds, starting with simple molecular multiplications [repetitions] and up to the multiple presence of invertebrate organs [in centipedes, worms, and insects], and even of vertebrate organs or structures, possibly express this compensation of disturbances ( NOTE 882).
The, for interactions superbly favorable, chemical bonding among all -- found and separated by us analytically, otherwise, however, always truly bonded already from the beginning -- partners is also an almost unavoidable precondition for functional differentiation needed for enhanced capability of performance, a differentiation which we first see in metazoic tissues and organs, and then were able to see them in protozoans [unicellular organisms] too, and which [differentiation] we must now trace back into the molecular range as the true point of departure.
The cellular, and ultimately the intra-cellular differentiation, is not an appearance of different structures out of a uniform base, but the unsymmetrically itself-enhancing expression of a -- with this becoming recognizable, but before this not yet visible -- polyvalence [i.e. a poly-potential substrate, in which thus the "new" structures are already present, but still in an enfolded state]. Living matter, of the type of the primordial living substance, owes its ability of sensibility, contractility, etc. from determined molecular-structural features, which alongside each other, or better, with one another, exist and have to exist, and of which some may be forced at the expense of others which now relatively strongly recede in their expression and performance (as to the same function) [This, then, is Müller's explanation of the above mentioned polyvalency.].
The stronger the differentiation, i.e. the greater the ability of performance of the changed substance becomes, the less self-able as to exist it [the differentiation] becomes, i.e. a once commenced differentiation has, in behalf of securing life and of a general competence to live, as consequence that it either has "to its disposal" a correspondingly large amount of toti-potent undifferentiated matter ( NOTE 883), or that also the other features are differentiatingly enhanced, through which a maximum of performance becomes possible with a minimum of mass. Both cases, but especially (namely in many stages and transitions) the last one, may be realized [in order for the strategy of the organism to be effective].
Functional differentiation known as division of labor cannot be realized at one and the same place or within a very narrowly confined molecular region at the same time. So here we have to do with a spatial distinction, which however, neither needs to be a spatial separation, nor can be it. [The whole remains intact]. In order to restore the indisputed functional original unity of a first living molecule, one needs one or another more or less complex connection element [connecting the workers, and/or connecting the instrument with its proper substrate, i.e. that what is treated], whose nature is, however, totally unknown to us. One can spare oneself this doubtful and toilsome course if one doesn't let disintegrate the unity ( NOTE 884) in the first place, but preserves it in [the form of] the intra-molecular connection. This means that the living substance remains in its true molecular bonding framework, and it does so independently of differentiation on the one hand, and quantitative extension on the other. The commenced differentiation does need the true bonding relation in the sequel, because its own ability to live has become decreased.
We again summarize : Interaction of molecules varies between the two extremes of (1) the Van der Waals attraction, and (2) the complete chemical transformation, that is to say in going from (1) to (2) : from an effect, usually indicated as dispersion, which [effect] is not localized at any determined points of the molecules (and thus being some sort of unspecific overall shell effect [spheres of minimal distance between particles, resulting from Van der Waals attraction] ), up to a fixed mutual coupling of the electronic systems [of the combining atoms]. The dispersion effects may be already rather strong. And although these attractions are [as to their strength] only fractions of chemical bonds, one might, in organismic substances, think of the possibility of extreme values assumed by these attractions, values reaching those of chemical resonance. But in this always remains -- viewed individually -- the uncertainty factor of the mere statistically average unification [of particles], and the very great susceptibility to disturbance of any kind (small range, steep decline with the thermally varying inter-molecular distance, etc. [rendering restoration of the initial unification unlikely] )
If one moves from this still more or less loose interaction towards the firmer connection in stoichiometric super-molecules [bonding partners in definite and constant proportions] or in molecular compounds (MC) (which also should be stable in "solution"), then one will find out that protein molecules, in addition to their evident capability to form salty compounds, also possess the most important constitutive properties rendering them capable of forming molecular compounds. Because still more possibilites exist, one may reckon that involvement of quantum-resonance phenomena results in rather strong properties of stabilization but now already too much establishing the frozen "non-living" state.
[As to the constitution of living substance] absolute secure and permanent inter-molecular forces are demanded (of whose significance in living substance one was always convinced, but which one also has treated as "free" forces), which [forces] not only are effective at shortest distances, but are effective unalteredly and evenly also at all temporally appearing distances. Such forces, having at the same time to be strong, are not known, and there is no evidence to be found in Nature that they must somehow occur. So the only way out of this dilemma is keeping the distance [between interacting particles] always evenly small, and unaltering. Such permanent proximity is certainly known, and the forces causing it are in no way peculiar. They are those most import forces of the chemical bond, forces holding together matter, forces appearing in their different forms, and with the unequivocal and central form of the covalent bond. There is no compelling reason to doubt that the chemical forces do also create the organismic constitution and coherence in its totality. So whereas the "pure" system doctrine is forced to preferentially or exclusively have [in the organismic constitution] involved cohesion effects as molecular residual fields (low-capable scatter field effects instead of the "toti-potent" electronic and nuclear fields), we put forward the undiminished chemical bond, and believe to have made probable its presence not merely based on the various consequences but also having it demonstrated practically effectively.
Only the chemical bondings within the molecule, and in our case within the organismic mega-molecule of arbitrary size, are sufficiently firm to resist the thermic molecular motion (note that the resistance of the molecule against stretching [and thus against snapping] is about ten times stronger than that against bending), whereas these thermic motions can effortless break up the "bonds" between the molecules. With this, we do not overlook that the inter-molecular "bond" always restitutes itself statistically (also "dynamically" as one likes to express things), and thus only at all exists statistically. But we have of organismic functioning -- not only of heredity -- the impression [of this functioning to be] of such an unshakable constancy, that we can place it nowhere else than under the most secure, namely under the chemical bonding relationship.
What makes us disloyal to the alleged system, and to the colloidal or crystalline order, of the living substance are, in addition to many other decisive arguments from the origin of life, the organismic ontogenesis [individual development], etc., the fundamentally important phenomena of organismic summation (namely of phenomena of life, and thus not, for instance, the summation of potentials in the electrical organ of certain fishes). In classical physics -- not considering the simple mass addition or additive effect of directed elementary magnets -- there doesn't exist a proper example of this summation. Ionic crystals [crystals made up of a lattice of ions] are already over short internal distances [i.e. already in very small crystals] electrically neutral as to their outwardly directed behavior (whereas along shorter internal distances in the order of 10-8 cm in the NaCl crystal lattice there occur, between the ions, field strengths of about 400 000 000 Volt per centimeter). The same holds for liquid dipoles [such as water molecules]. And inter-molecular field effects (compare proton motility) either remain isolated = locally confined [thus no summation], or they are large-range effective through molecular chains as alternating inductive effects [these are thus merely passed on], here, in special cases with a certain degree of strengthening (additivity), it is true, but still with a limiting value, still lying below the idealized total-polarizing of a single "key atom" [in the conduction chain, and thus also no summation]. The same is seen in all models of electronic or protonic energy conduction. Here it is superfluous to emphasize that it is here not about one or another pecularity, but that such effects, on quantum mechanical grounds of resonance stability, can only be such as they are actually found to be in practice. With thought models one may, it is true, obtain certain extremes, but may not go beyond the micro framework. As to living substance, one would -- in this way -- think of an example-lacking structure, or (which would then be better) assume that "something" of a quite different nature, someting alien to known classical physics, is summed up and powered. And in order for that to be possible, one should cling especially to this exampleless or modelless structure.
While the above physical and chemical systems are always characterized by a regular alternation -- and thus a determined schematical order -- but the protoplasmatic substance, according to the results of research in amino acid sequence, being built up contra-periodically (we hold "aperiodic" to be too "weak" an expression), one could -- as a partial element at least -- hold the lack of order (of the type known to us) in organisms to be responsible for the fact that here [in organisms] no demise as a result of neutralizing [as the outwardly non-effectiveness of electrical charge in a NaCl crystal], saturation, and short circuit takes place, while the possibility of summation remains open. With this, one is precisely outside the known physical, because there the non-orderly (= chaotic) has the true and example-giving minimal opportunity of any additivity-of-effect or of intensity-increase. The chaotic, which simply is a disorder, is as much opposed to the anti-order of the living as is the full ordering of the crystalline dead.
[Indeed, the disorder, the chaotic, in the physical domain, i.e. a random structure of things, has no possibilities for summation or addition (= approximate repetition = strengthening = intensification), because, as a result of the randomness, it has no distinct subregions, subregions that profile themselves against other such regions, regions that are, in the structure, approximately repeated, or can be so repeated.
On the other hand, the non-order in the organismic domain, as it is in the structures of organisms, is such that such a structure does not exclude the possibility of summation or addition, i.e. of approximate repetition, of strengthening, of intensification, because this non-order here means, not disorder, not randomness, but merely contra-periodic structure. It is avoiding periodicity, without becoming by that reason a random structure. And although true disorder is also not periodic, it is not disorder merely as a result of lacking any periodicity, but is disorder as a result of the randomness of the structure.
Organismic structure, i.e. the structure of the organismic mega-molecule, is contra-periodic because it must embody some message. This message is about the function or functions of the molecule, like the amino acid sequence in an enzyme molecule determines the three-dimensional shape of that molecule and, as a result, determines (or encodes for) its function. A periodic structure, on the other hand, cannot encode any message, and although also a random structure cannot apparently encode any message it might turn out to be a message or a jumble of fragments of some message. So in contrast to a random structure, a periodic structure, as we see it in crystals, precludes any possibility to be able to carry a message, and therefore the organismic structure is not merely a-periodic, but explicitly contra-periodic.
To elaborate on periodicity still further : In Fourth Part of Website we investigated whether there is an analogy between crystals and organisms, and we found out that there is such an analogy, especially evident in dendritic snow crystals (and then, of course, also in all other dendritic crystals). But we didn't find any true and clear lattice structure in organisms. So we must now say that we, after the fact, didn't look for such an analogy at all, but only in what degree there might be such an analogy. And indeed, there is some degree of analogy as can be seen in the features (considered in their generality) of growth, regeneration, specific shape, and the far-from-thermodynamic-equilibrium state, all present in both (dendritic) crystals and in organisms. But now we see that the analogies stop precisely where we have found them and in what condition we have found them to be : growth and regeneration (and some other analogies) as they are and take place in crystals, i.e. as they are supported by periodicity of structure, cannot be further developed. They are end-points, they are dead-end pathways towards true growth and regeneration. They cannot become truly sophisticated as they are in most organisms. And this is, among other things, because encoding and prescription are impossible in and by a periodic structure. So the "crystal analogy", as investigation, must develop into a "crystal antilogy" demonstrating the fact that periodicity, crystallinity, blocks the path toward life. Of course crystalline substructures do occur in organisms, but they are just non-living assets and parts. Truly living organismic substance is non-crystalline and carries the message of its function.]
In the next document we continue with Müller's exposition [with explanation and additions of mine] on Unimol.
e-mail :
( Please write in ' Subject ' entry : ' METAPHYSICS ', in order for me to be able to distinguish your mail from spam )
To continue click HERE for further study of the Unimol View of organisms, rendering organisms to be homogeneously (metaphysically) classified with all other molecular Substances, Part XVi.
Back to Homepage ( First Part of Website)
Back to first part of theoretic intermezzo
Back to second part of theoretic intermezzo Back to Part IX
Back to Part XIV, Dipterygia, and conclusion of flight-devices in insects
Back to Part XV, first part of the theory of inorganic nature
Back to Part XVa, second part of the theory of inorganic nature
Back to Part XVb, third part of the theory of inorganic nature
Back to Part XVc, fourth part of the theory of inorganic nature
Back to Part XVd, fifth part of the theory of inorganic nature
Back to Part XVe, sixth part of the theory of inorganic nature
Back to Part XVf, seventh and concluding part of the theory of inorganic nature
Back to Part XVg, Unimol, theory of the organism as one single molecule (first part)